【7.2】活性药物发现项目中结合自由能计算的大规模评估
使用计算方法就化合物与蛋白质的结合亲和力对化合物进行准确排名,对药物研究非常感兴趣。基于物理的自由能计算被认为是估计结合亲和力的最严格方法。近年来,在学术界和工业界进行的许多回顾性研究都证明了其潜力。在这里,我们展示了FEP +方法在德国达姆施塔特默克(Merck KGaA)行业环境中的活性药物发现项目中大规模前瞻性应用的结果。我们将这些前瞻性数据与在八个药物相关靶标的新的,多样化的公共基准上获得的结果进行比较。我们的结果提供了对在现实生活中的药物发现项目中使用自由能计算时面临的挑战的见解,并确定了未来方法开发可以解决的局限性。我们提供给社区的新的公共数据集可以支持进一步的方法开发和自由能计算的比较基准。

一、前言
小分子药物开发项目的目标是,在与其他与安全性和生物学功效相关的配体特性保持平衡的同时,鉴定与目标蛋白具有高亲和力的配体。 为了支持这一具有挑战性的企业,长期以来,准确预测蛋白质与配体结合的自由能一直是计算机辅助药物设计(CADD)的目标。 使用蒙特卡洛或分子动力学模拟的基于物理学的自由能计算被认为是解决此问题的最严格方法。 然而,直到最近,高昂的计算成本,采样算法的局限性以及(小分子)力场的局限性阻碍了广泛的使用。 此外,从行业的角度来看,由于协议的自动化,吞吐量和健壮性有限,因此使用自由能计算被认为具有挑战性。
然而,在过去的十年中,在采样,(1-8)力场开发,(9-18)吞吐量,(19-28)和自动化方面取得了巨大进步。(5,14,29-41)结果,许多制药公司采用了相对自由能计算(42),尤其是SchrödingerFEP +工作流程(5)作为支持其药物发现工作的新计算工具。 2016年,我们在德国达姆施塔特(Darmstadt)的默克公司(Merck KGaA)发起了一项大型计划,以全面评估预测准确性并确定自由能计算的最佳用例。在这项计划中,我们旨在将FEP +应用于所有合适的内部主动药物发现项目。在现实的预期应用程序中测试新方法的这种特殊设置的三个主要原因。
- 首先,已经证明了对计算工具的盲目评估,例如以社区广泛的预测挑战的形式,例如CASP,(43)CAPRI,(44)SAMPL,(45)和D3R Grand Challenges(46),可以提供更现实的结果给定方法的准确性和局限性的图片。因此,在这项计划中,我们专注于前瞻性地(即盲目地)应用FEP +。
- 其次,目前尚不清楚在现实生活中的药物发现项目中普遍存在的时间限制,资源限制和信息鸿沟相对于先前文献中报道的方法会影响该方法的性能。(5,17,47, 48)
- 第三,自2015年对FEP +方法进行了大量初步评估以来,(5)该方法增加了许多功能,例如计算涉及开环和净电荷变化的转化的自由能。(47,48)这些挑战转化类型通常用在药物开发项目中,但以前的基准数据集中却没有。
在这里,我们介绍了过去三年中在18个内部药物发现项目中收集的回顾性和前瞻性数据。 我们描述了为在项目中使用自由能计算而建立的一般工作流程,并报告了内部目标上FEP +方法的性能。 我们进一步介绍了从新的多样化基准中收集的数据,并讨论了适用范围,项目影响和方法局限性的关键知识。
二、结果与讨论
自由能计算,特别是Schrödinger的FEP +实施,已经成为一种精确的结合亲和力预测方法,可以潜在地加速从候选物到候选物的优化。尽管该方法的理论精度取决于所用力场的精度和采样的范围,但实际上,系统设置和结构准备也会影响性能。因此,尚不清楚在行业设置的约束条件下应用该方法时,能否保持以前报告的方法(5)的准确性,以及如何最好地为化合物设计和相关的多参数优化过程做出贡献。因此,2016年,在德国达姆施塔特(Darmstadt)的默克(Merck)股份公司(Merck KGaA)发起了一项大型计划,以彻底评估FEP +技术并在未来的项目中对其进行基准测试。从2016年到2019年,我们将FEP +应用于12个目标和23个化学序列,进行了超过35,000个单独的扰动计算。我们最终获得了针对6,000多个化学实体的有效预测。合成和测试了400多个盲目预测和新颖的分子。这产生了大量的前瞻性数据,提供了在典型的小分子药物发现工作环境中对该方法准确性的详细评估。除基准测试外,该计划还使我们能够定义最佳实践并探索最佳用例。
2.1 项目中的自由能计算工作流程 Free Energy Calculation Workflow in Projects
在过去三年中,我们建立了用于在项目中部署自由能源计算的工作流。该过程如图1所示。首先,我们通过收集可用的结构数据和实验确定的结合亲和力,评估将FEP用于给定目标和感兴趣的化学系列的一般可行性。在这一阶段,我们通常需要至少一个分辨率良好的共晶体结构,其中一个代表感兴趣的系列。严格的要求是基于我们在三个项目中的经验,在三个项目中我们尝试在没有X射线结构的情况下使用同源性模型。与以前使用同源模型的FEP成功应用相反,(49-51)我们回顾性地在所有三种情况下(RMSE> 1.5 kcal / mol)都获得了令人满意的预测准确性。在这三个项目中的两个中,我们后来获得了共晶结构,然后能够成功完成验证阶段(请参阅“内部药物发现项目中的FEP可行性和验证结果”)。虽然我们没有成功地将同源性模型用于FEP,但是对于结合模式假设,我们有混杂(mixed)的经验,该假设是通过在不存在感兴趣系列的共晶结构的情况下对接而获得的。在FEP评估过程中,我们在两个项目中遇到了这种情况。在这两种情况下,我们都尝试根据在第一种情况下使用另一系列的共晶体结构和在第二种情况下蛋白质的载脂蛋白结构获得的对接位姿进行验证研究。在第一个项目中,我们无法与实验数据取得良好的一致性。后来获得的共晶体结构在结合位点显示出相当大的蛋白质柔性,尽管预测的结合姿势与晶体结构相对相似。在第二个项目中,我们确实能够使用对接姿势作为模型系统在预测和实验亲和力之间获得良好的一致性(图S1(A))。后来,我们使用了同时解决的共晶结构进行了第二次验证研究。这证实了对更大和结构更多样化的一组配体的良好预测准确性(图S1(B))。对接姿势与共晶结构所示的结合模式非常相似。

一旦获得足够的结构数据,我们将收集具有实验结合亲和力(至少10个配体,最好为20个)的同类配体的数据集,以及有关生化和生物物理测定的所有可用信息(例如蛋白质构建体,缓冲液条件,辅助因子等)。请注意,配体数据集的大小建议不是“经验法则”,通常很难在早期项目中获得更大的数据集。如果配体数据集足够大,则可以根据分子的R-基团进行拆分,对这些R-基团进行修饰,以识别结合位点不同部分中预测的潜在准确性。然后基于这些数据集,进行回顾性自由能计算,以便将预测的结合亲和力与实验确定的结合亲和力进行比较。我们通常将这些回顾性计算实验称为验证研究。通过计算集合中所有配体对的相对预测亲和力ΔΔGpred和相对实验亲和力ΔΔGexp之间的成对均方根误差(RMSEpw),可以评估自由能计算的准确性。 ΔΔGpred是通过减去预测值ΔGpred(包括周期闭合校正)来计算的。如果可以,将预测的亲和力与不同测定的实验结果进行比较。通常,在验证阶段,将评估不同的输入结构和设置,以便找到最佳设置以用于以后的预期计算。在实践中,时间限制通常将这一阶段限制为通常评估3种可能的模型系统。模型系统在输入结构,采样时间和采样设置(例如λ窗口的数量)方面可能会有所不同。如果已经解决了蛋白质的多种结构和/或目标序列,输入结构的变化可能涉及所用的晶体结构,结合模式(不同的对接位姿或与电子密度兼容的不同位姿),蛋白质的一部分使用的结构(单域或多个域,如果适用),如果在晶体结构中观察到的低聚物具有已知的生物学功能,则该蛋白质的低聚状态,或者是否考虑了目标蛋白质的可能的蛋白质-蛋白质相互作用伙伴
对于较大的异常值(|ΔGpred–ΔGexp|> 2 kcal / mol),将进行详细分析以了解潜在原因。 如果我们获得的RMSEpw小于1.3 kcal / mol,并且如果我们能够充分解释较大的异常值(如果存在),我们通常认为验证研究是成功的。 尽管FEP所需的确切准确度取决于应用,通常认为最大2 kcal / mol的准确度对于对大型文库进行评分是有用的(52),但我们发现在验证研究中,RMSEpw的严格阈值<1.3 kcal / mol给出了 我们有更好的机会在预期环境中获得足够的精度(RMSE <2 kcal / mol)。 在生产模式下,与验证研究相比,预测准确性通常会下降(请参见下文)。
如果数据集的动态范围合适,则FEP预测也应产生良好的排名。 然而,实际上,我们经常遇到这样的情况:在验证研究时,只有动态范围有限的数据集可用,这使得模型评估具有挑战性。(53)在这种情况下,我们进入了预期阶段。 一种“试用”方式。 我们在不使用优先级预测的情况下预测了所有正在合成的分子,并评估了这些前瞻性预测以评估该方法的准确性。 根据这些结果,我们决定是否在生产模式下将FEP应用于该项目。
成功完成验证阶段后,FEP项目将进入生产模式,并对复合构想进行前瞻性计算。这些想法必须与经过验证的化学系列足够相似。对于新的化学系列和新的可用结构信息,必须进行新的验证研究。我们在整个项目中密切监视预测的预期准确性,并跟踪已合成的预测化合物。所有预测的亲和力和结构都存储在数据库中。使用自动化的工作流程,我们会定期检查内部数据库,以检测在此期间是否已合成并测试了预测的化合物。根据我们的经验,这种对合成分子的持续监控以及由此而来的预期预测准确性对于在项目团队中最初建立信任并随后检测化学物质何时移出模型的适用范围是必要的。首先根据精确的结构匹配进行结构匹配。如果未找到匹配,则基于匹配的互变异构体或匹配进行匹配,而无需考虑立体化学以最大化收集的数据数量。将指定“匹配模式”的标签分配给最终结果,并手动检查所有基于互变异构体或不考虑立体化学而匹配的数据。在有多种异构体的情况下,一个预测要与数据库中的多个条目和活动相匹配,并且必须通过视觉检查来分配正确的预测。我们假定与自由能方法的典型误差相比,将单个异构体的预测值与各自的外消旋物匹配不会导致较大的误差。但是,很明显,这可能并非对所有项目都有效,因此在分析预测准确性时应考虑这些假设。尽管如此,对于我们的数据集,最大的预期异常值并不包含任何仅以外消旋体形式合成的,具有确定立体化学的分子(请参阅“内部项目中预期的FEP +结果”)。
2.2 内部药物开发项目中的FEP可行性和验证结果 FEP Feasibility and Validation Results in in-House Drug Discovery Projects
在三年的过程中,我们评估了28个总体FEP可行性目标(图2(A))。我们对18个目标和44个化学系列进行了验证研究,并对14个目标和25个化学系列进行了前瞻性计算(如前所述,起初,我们认为验证成功的标准更为宽松)。注意,我们定义了项目中使用的化学系列;但是,这是特定于项目的,通常取决于所涉及的化学家。无法针对给定目标执行验证研究的主要原因是缺乏相关的结构数据(7个目标)。对于三个目标,所有验证研究都被认为是不成功的;对于另外两个目标,在完成FEP可行性评估或验证研究后(项目组合类别),该项目立即中止。尽管获得了良好的验证结果,但某些化学系列产品仍未进入生产模式,因为由于其他原因,项目团队同时取消了它们的优先级。总体而言,一旦有足够的结构和结合亲和力数据可用于进行验证研究,我们观察到潜在FEP靶标的损耗率相对较低。

图2(B)显示了针对18个目标的验证研究中实现的精度。总的来说,我们能够获得14个目标和21个化学系列的高精度(RMSEpw <1 kcal / mol)和可接受的精度(RMSEpw <1.3 kcal / mol)预测。在该计划的早期阶段,我们还接受了RMSEpw大于1.3 kcal / mol的验证研究成功,因此使这些系列进入了生产模式。在我们的评估过程中,我们收紧了成功进行验证研究的标准,因为验证研究中较低的准确性(RMSEpw在1.3至1.6 kcal / mol之间)始终导致预期环境中的准确性甚至更低。预测精度不仅在不同目标之间而且在同一目标蛋白的不同化学系列之间也不同。最值得注意的是,对于一个目标,我们获得了多个化学系列的结果,涵盖从高精度(RMSEpw <1 kcal / mol)到低精度预测(RMSEpw> 2 kcal / mol)的整个范围(图2(B),浅粉红色)颜色)。此外,在活动项目中使用FEP时,我们经常会遇到可能影响方法准确性的挑战。对这些挑战的定性评估如图2(C)所示。几乎所有项目都有至少一个方面可能对应用自由能计算有问题(图2(C)):毫无疑问,现实生活中的药物发现项目并不是理想的案例。在我们尝试进行验证研究的项目中,我们遇到的最常见挑战是至少一部分配体的结合模式存在不确定性,并且由于怀疑的蛋白质构象变化而导致蛋白质结构存在不确定性(分别为66%和44%) 。在六个项目中,我们发现实验数据的来源会影响我们是否可以认为验证研究成功。在一种情况下,我们最初将预测的亲和力与功能分析的输出进行了比较,发现偏差较大。但是,当将相同的预测亲和力与生物物理表面等离子体共振(SPR)数据进行比较时,我们发现了很好的一致性,因此决定将系列推向生产模式(图S2)。一方面,可以预期FEP结果通常应与生物物理结合数据最匹配,并且与其他类型的测定法相比,可能会显示更大的误差。另一方面,要对项目产生重大影响,FEP预测的亲和力应与用于驱动化合物优化的主要分析方法密切相关。而且,在许多项目中,与来自生物化学或功能测定的数据相比,仅有数量有限的生物物理数据可用。我们不鼓励在主要优化分析与生物物理数据没有很好关联的项目中进行投资来进行验证研究,因为FEP对项目的影响可能仍然有限
在四个项目中,我们发现小分子力场的参数可能无法准确描述相互作用。在其中两个项目中,使用更高版本的OPLS3e力场提高了验证研究中使用的配体组的准确性(数据未显示)。在一个项目中,力场的变化与部分装药有关。在第二个项目中,该化合物具有取代的脂族环。如最近版本的“力场生成器”中所做的那样,该环中扭转电势的重新参数化有助于提高预测的准确性。对于这三个项目,我们发现了较大的离群值以进行前瞻性预测;但是,使用较新版本的力场进行重新计算不会更改结果(以下情况将讨论这些情况)。
请注意,这里我们列出了可能影响预测准确性的潜在方面。但是,我们通常无法将验证研究的成功概率确定为特定功能。例如,我们分析了X射线结构的分辨率是否对验证研究中获得的准确性有影响,但没有发现相关性(数据未显示)。
2.3 内部项目的预期FEP +结果
对于12个目标和19个化学系列,可以获得包含至少五个数据点的前瞻性数据集。 每个目标和系列的结果如图3所示(另请参见表S1,了解通过对每个数据集进行自举抽样计算得出的其他性能指标和置信区间)。 我们将效果指标报告为xxlowxhigh,其中x是从完整数据集(例如,RMSE)计算出的平均统计量,xlow和xhigh表示90%的置信区间(请参见实验部分)。

与验证研究的结果相比,我们发现较高的RMSE值表明准应用的准确性较低。请注意,此处我们通过比较每种化合物的预测自由能ΔGpred与实验亲和力ΔGexp而不是像验证研究中那样计算所有配体对的相对自由能ΔΔG的误差来测量准确性。原因是配体不是源自单个FEP图,而是源自多个独立的运行,在某些情况下,不同的分子被用作参考结构。在整个项目中使用不同参考分子的主要原因有两个。
- 首先,我们想尽可能地靠近用FEP筛选的新化学空间,以避免大的转化,并可能使该方法具有更好的性能。
- 第二个原因是可操作的:我们希望包括当时化学家最感兴趣的分子。
注意,使用不同的实验参考点可能对相对自由能方法的准确性评估产生问题。对于不同参比分子覆盖很大不同亲和力范围的情况,这尤其重要。例如,对于分别使用具有高亲和力和低亲和力的参比分子计算的两个图,根据参比分子,这些图的相对排名将是完美的(如果预测的自由能相对于该分子不显示大的变化)参考分子)。在这种情况下,对完整数据集的评估将导致对预测准确性的高估。但是,对于我们的情况,即使使用的参比分子也不相同,其亲和力范围也相似。
对于三个(尽管很小)的数据集,我们获得了高准确度的预测(RMSE <1 kcal / mol),对于四个数据集,我们获得了中度精度(RMSE <1.5 kcal / mol;这是我们期望看到的一系列数据的准确性)在验证研究中显示RMSE <1.3 kcal / mol),对于另外八组,我们获得了可接受的准确度预测(RMSE <2 kcal / mol)。具有中等准确度(RMSE <2 kcal / mol)的结合亲和力预测方法被认为可用于对较大的文库进行评分。(52)从我们的预期应用中,我们能够在19个化学系列中的17个中达到这一中等准确度水平。对于19组中的13组,我们发现相关性好(Kendallτ> 0.5)或误差低(RMSE <1.5 kcal / mol)。在图3中,目标按时间顺序排列。因此,该图还显示了一条“ FEP学习曲线”:前五个目标的RMSE高于最近五个数据集的RMSE(324个配体的RMSE = 1.781.42.12 kcal / mol,RMSE = 1.350.971.72 kcal / mol分别代表118个配体)。但是,由于置信区间较大,因此很难对改进进行清晰的评估。
我们调查了数据集中最大异常值的原因。对于分析,我们将所有预计在测定的顶部之上或之下的化合物的预测亲和力重置为这些值,然后评估与实验值的偏差。基于此分析,我们仍然确定了23种化合物,其中预测的亲和力与实验亲和力相差3 kcal / mol以上。对于源自靶标1 /系列1的这23个离群值中的四个,验证研究先前显示了该分子这一部分进行修饰的较大误差。根据FEP计划稍后建立的更严格的标准,我们不会将该项目推进到预期阶段。靶标1 /系列2和靶标1 /系列3中的另外四种化合物相对于参考化合物显示出芳族杂环的变化。最初,我们使用OPLS3力场,与实验的偏差甚至更大(绝对误差> 5 kcal / mol)。当使用OPLS3e力场重新运行这些力场时,该力场降低至≈3kcal / mol。我们还使用Schrödinger套件版本2020-1重新计算了目标1 /系列2中的化合物;但是,这并没有改变结果(所得到的最小示例图参见图S3)。虽然在该方法的典型误差范围内捕获了两种化合物之间的相对亲和力,但明显低估了这两种化合物相对于第三个支架的亲和力。
对于靶标2 /系列1和靶标5 /系列1中的三种化合物,我们怀疑相对于共结晶分子的变化可能引起了在模拟中未捕获的蛋白质构象变化(该变化是在部分在可用晶体结构中显示灵活性的结合位点)。对于来自目标5 /系列1的五个异常值的一组,当对分子的溶剂暴露侧进行修饰时,不是结晶构建体一部分的其他域可能导致效能突然下降(发现了五个分子尽管与高活性分子相比具有相对较小的修饰,但在测定中仍无活性)。对于晶体结构,使用仅包含具有活性位点的结构域的构建体(覆盖全长蛋白质的约三分之一)。相反,结合亲和力测定法使用全长蛋白质。所有这五个异常分子均被FEP高估了5 kcal / mol以上,从而对该数据集观察到的总RMSE为2.161.632.64 kcal / mol做出了重大贡献。当排除这五个分子时,对其余83种化合物计算出的RMSE降低至1.381.121.66 kcal / mol。
最后,源自目标8和目标9的两个异常值对砜或磺酰胺进行了修饰。当我们最初对这些分子进行FEP +计算时,我们注意到在替换砜或磺酰胺基团(当时似乎已经“可疑”)时,预计会有许多修饰是有利的。确实,当我们在这两个项目中合成最佳预测化合物时,我们发现亲和力被高估了4 kcal / mol以上。对目标8重复此计算,并在Schrödinger版本2020-1中使用最小的示例图重复进行,仍然显示出此高估(数据未显示)。我们还在针对HIF2-α数据集的公开基准中发现了类似情况(转换结果如图S4(A)所示)。这些示例可能暗示了OPLS3 / OPLS3e小分子力场在描述相对疏水的结合口袋中的砜的特性时存在问题。最近,Jorgensen及其同事提出了对OPLS力场中的固定电荷模型的改进,以更好地捕获含硫分子的性质。(54)但是,可能有必要包括极化能力以完全捕获此类含硫基团的蛋白质-配体相互作用
此外,与验证研究相比,有几个一般原因可以解释预期数据集中较大的误差。
-
首先,对于多个目标,预期数据集大于原始验证集。较大的样本量导致对RMSE的估计更加可靠。
-
其次,在整个项目过程中,新想法往往与验证研究中使用的化学空间不太相似,而与晶体结构中捕获的化学系列的代表则不太相似。这导致在配体的结合方式和质子化状态方面更高的不确定性。如果选择了错误的输入姿势并限制了采样,这些不确定性可能反过来影响计算的准确性。仔细准备输入结构和良好的3D对齐对于使用FEP +获得准确的自由能估算值很重要,因为自动地图生成依赖于输入结构。最初,我们使用柔性配体比对将新型分子置于结合位点。但是,忽略蛋白质有时会产生次优的结构。在我们手中,使用受MCS约束或自定义SMARTS约束的Glide可以提供更好的起始姿势,而无需手动调整姿势(这在行业环境中也很重要)。这与最近的出版物一致,该出版物系统地研究了不同的比对方法对FEP +预测的影响。(56)在许多情况下,初始比对后,结合方式仍然不确定,例如,不对称取代的环。在这种情况下,以及由于互变异构体或电荷状态的不确定性,我们在对全套化合物进行排名时,要么在图中模拟了所有可能的状态。或者,我们首先尝试用FEP +(结合姿势FEP)预测相关状态,然后根据每种化合物的预测最佳状态对整个集合进行排名。最近,有可能在FEP +中包含多个质子化状态。(57)但是,在蛋白质结合后改变质子化状态的情况下,或者配体在结合位点与溶剂中具有不同的pKa的情况下(58) -60)仍无法正确处理。在过去的十年中,恒定pH模拟取得了长足的进步(61-66),但目前,这些方法在计算上似乎对工业环境要求很高。
-
第三,我们发现内部化合物集合中的各种化学物质仍然是小分子力场的挑战。对于几乎每个新的化学系列,即使使用了具有非常高的扭转电势的新型OPLS3e力场,我们也必须重新设定一些扭转电势。(17)诸如SMIRNOFF的“ Parsley”力场等新概念由Open Force Field Consortium(67)在帮助更轻松地扩展新颖化学的力场和确定合适的参数方面似乎很有前途。我们预计,未来小分子力场将继续改善并更好地捕获不同化学基团的特性。反过来,这将有望减少自由能计算中的误差。
-
第四,在前瞻性地使用自由能计算时,我们倾向于关注极端预测(例如,排名最高的化合物)。可以通过例如应用选择偏差校正来减轻针对极端预测的偏见。(68)但是,在我们看来,这并没有对最大的异常值产生太大影响。
-
最后,项目团队非常感兴趣的许多转换都对该方法产生了固有的挑战(例如,从芳香环系统到脂肪链,电荷变化,通过柔性连接子添加新基团)。尤其是,在采样方面很难添加具有灵活连接子的新功能组,但是它经常发生在早期命中优化和片段优化的背景下。
将FEP +的排名与从Glide停靠,Glide计分和Prime分子力学广义Born表面积(MMGB-SA, molecular mechanics generalized Born surface area)评分获得的排名进行比较时(请参阅实验部分以了解详细信息),我们发现整体FEP +的性能要优于这些标准结构- 基于药物的设计(SBDD)方法(表S2)。 肯德尔τ的Cohen d与Glide对接比较为0.85,Glide对射得分为0.67,Prime为0.58,表明效果大小中等。(69)请注意,与Glide对战和Prime相比,目标6 /系列1为 未考虑用于计算效应大小,也出于技术原因,无法使用其他两种方法对所有配体进行排名(例如,某些配体因对接而无法对接蛋白质或使其最小化,或者在Prime MM-GBSA中给出阳性评分 因此被排除在分析之外)
在四种情况下(目标1 /系列4,目标4 /系列1,目标5 /系列3和目标6 /系列1),Prime MM-GBSA似乎产生了相似或更高相关性的良好排名(Kendallτ> 0.5)。比基于FEP +的排名要高。在其中两种情况下(目标1 /系列4和目标4 /系列1),对于Glidescore也是如此。但是,由于数据集都非常小(<10个配体),因此置信区间非常大,因此很难就不同方法的相对性能得出最终结论。尽管如此,这可能仍暗示着有机会在某些情况下使用更简单的评分方法,并以像FEP +这样的计算昂贵的方法来关注其他方法无法对配体进行排名的情况。
我们还将前瞻性内部数据集上的FEP +性能与通过简单的描述符(例如分子量和log P)进行排名进行比较(相关统计数据在表S3中显示)。 FEP +也优于这些“无效模型”,如大而中的效应大小所表明的(肯德尔τ的Cohen d = 1.03,而FEP +的分子量和log P的比较则为0.64)。(69)glide 对接表明效应大小较小在分子量上的排名并不比对数P的排名好(Kendallτ的Cohen d分别为0.36和0.06)。glide 计分显示出较小的效应值(肯德尔τ的Cohen d分别为0.47和0.21),Prime MM-GBSA得分显示了中等大小和较小的效应量(Kendallτ的Cohen’s d分别为0.57和0.2)。(70)
2.4 基准结果 Benchmark Results
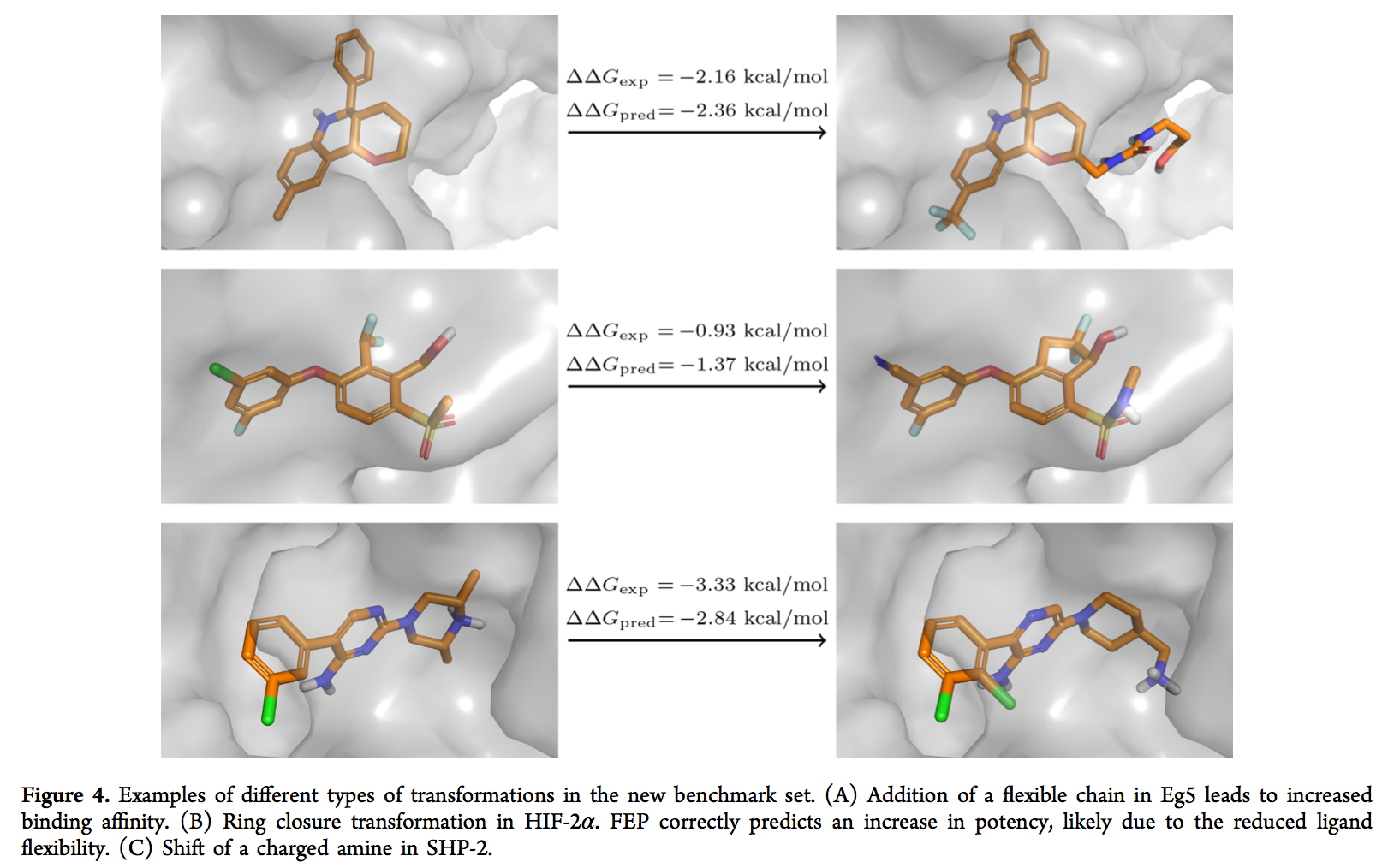
基于我们在内部项目中自由能量计算的丰富经验,不幸的是我们目前无法发布结构,因此我们决定构建一个新的基准,该基准由八个具有挑战性的,最近发布的,具有药学相关目标的数据集组成。(71− 80)基准总共包含264个配体。此基准数据集中的蛋白质目标和化学空间代表了我们内部项目中的目标。但是,配体系列与内部数据集没有重叠。炼金术转化的类型和大小反映了lead到lead和lead优化过程中化学变化的类型和大小。总体而言,此数据集很好地说明了我们在项目中面临的许多挑战。与先前发布的基准相反,(5)转换包括分子的净电荷和电荷分布以及开环和核跳跃的变化(示例如图4所示)。与以前的基准相比,配体组还显示出结构多样性的范围略有增加(使用默认设置的RDKit Daylight指纹,平均最大平均成对田本相似度为0.790.680.87对0.840.760.92)。 FEP +结果总结在表1中(每组的详细图可在图5中找到)。总体而言,我们为这些数据集实现了良好的相关性(平均值R2 = 0.440.250.64,平均值Kendallτ= 0.500.330.64)。在图RMSEpw中所有配体对上计算的相对亲和力ΔΔG的均方根误差在1.2和2.1 kcal / mol之间。请注意,与使用ΔG的预期数据集相比,此处使用ΔΔG来评估RMSE。原因是FEP +使用所有实验自由能ΔGexp来计算用于推导预测亲和力ΔGpred,(5)的偏移量,我们希望避免对性能进行高估。鉴于此数据集中包含的挑战,这是一项了不起的成就。但是,与先前在大型基准测试集(5)上报告的准确性以及Schrödinger的内部药物发现项目(RMSE = 1.1 kcal / mol,Lingle Wang,个人交流)相比,我们看到的性能要差得多。该较低的准确度与我们内部项目中发现的预期准确度一致(分别为平均RMSEpw = 1.681.591.76 kcal / mol和平均RMSE = 1.641.261.97 kcal / mol)。分析不同类型转化的误差时(图S5),我们发现涉及净电荷或电荷位置变化或分子核心/骨架变化的转化显示出较低的准确性。 FEP +软件已经认为这些转换固有地更加困难,并且使用了特殊的采样设置。(47,48)尽管如此,这些转换仍显示出较大的误差。有趣的是,对于其余的转换,我们无法观察到误差对转换大小(即被改变的重原子数)的强烈依赖性。 Christ and Fox在较早的评估中也意识到了这一点。(36)虽然乍看起来似乎违反直觉,但这可能反映出以下事实:被扰动的重原子数不能精确地衡量给定原子的“难度”。转型。但是,定义更好的措施是一项超出本研究范围的工作。


有趣且令人震惊的是,根据我们成功进行验证研究的指导原则(良好的相关性和RMSEpw <1.3 kcal / mol),我们只能在生产基准中的八个化学系列中进行一个 模式基于默认设置为5 ns采样时间获得的数据。 每个λ窗口将仿真时间从5 ns增加到20 ns,会使八个目标中的七个目标的RMSEpw降低0.05 kcal / mol以上,平均RMSEpw降低0.13 kcal / mol(平均RMSEpw = 1.511.441.73 kcal / mol, 参见表S4)。 然而,预测亲和力和实验亲和力之间的定量一致性的增加对排名/相关性没有影响(平均肯德尔τ= 0.490.320.65)。 但是,基于20 ns的数据,我们将能够将八个目标中的三个推进到生产模式。
为了调查FEP +的最新版本是否在基准上显示出改进的性能,我们使用Schrödinger2020-1版重新计算了PFKFB3和SYK数据集。 对于PFKFB3(τ= 0.640.540.73,RMSEpw = 1.691.631.75 kcal / mol),我们获得了稍微改善的性能,对于SYK(τ= 0.300.130.46,RMSEpw = 2.152.082.22 kcal / mol),获得了较低的精度。 因此,在FEP +(81)中引入诸如GCMC水采样之类的功能并不能解决在此基准上看到的所有挑战,因此可能需要进一步的工作。
我们将更详细地讨论c-Met和SYK FEP +结果。 c-Met数据集的准确性适中(RMSEpw = 1.431.341.51 kcal / mol),但与实验亲和力相比,预测的ΔG值显示出良好的相关性和排名(图6(A))。 ΔΔG的误差似乎遵循高斯分布(图6(B))。数据集包含带有溶剂暴露的碱性基团的12种中性化合物和12种带正电荷的化合物。 FEP图包含六个扰动,其中涉及净电荷的变化。对于其中的五个转换,预测的ΔΔGpred在实验值的1.1 kcal / mol以内(示例如图6(C)所示)。剩余的电荷变化转化涉及分子CHEMBL3402762,其分子的亲和力IC50小于1 nM(测定的顶部,将其设置为1 nM,以便在比较中包括该化合物)。预计该化合物比报道的IC50 = 1 nM的CHEMBL3402761更有效。总体而言,c-Met数据集中的扰动表明,可以通过FEP +方法可靠地处理涉及净电荷变化的转换。尽管如此,基准案例还说明了预测电荷变化所涉及的挑战。在TNKS2组中,准确预测了具有相同净电荷的配体内的相对亲和力(例如,对于所有21种中性配体,RMSEpw = 1.11 1.03 1.18 kcal / mol)。但是,不同净电荷的配体之间的相对亲和力显示出较大的误差,导致由27个配体组成的整个数据集的RMSEpw = 2.20 2.08 2.32 kcal / mol。
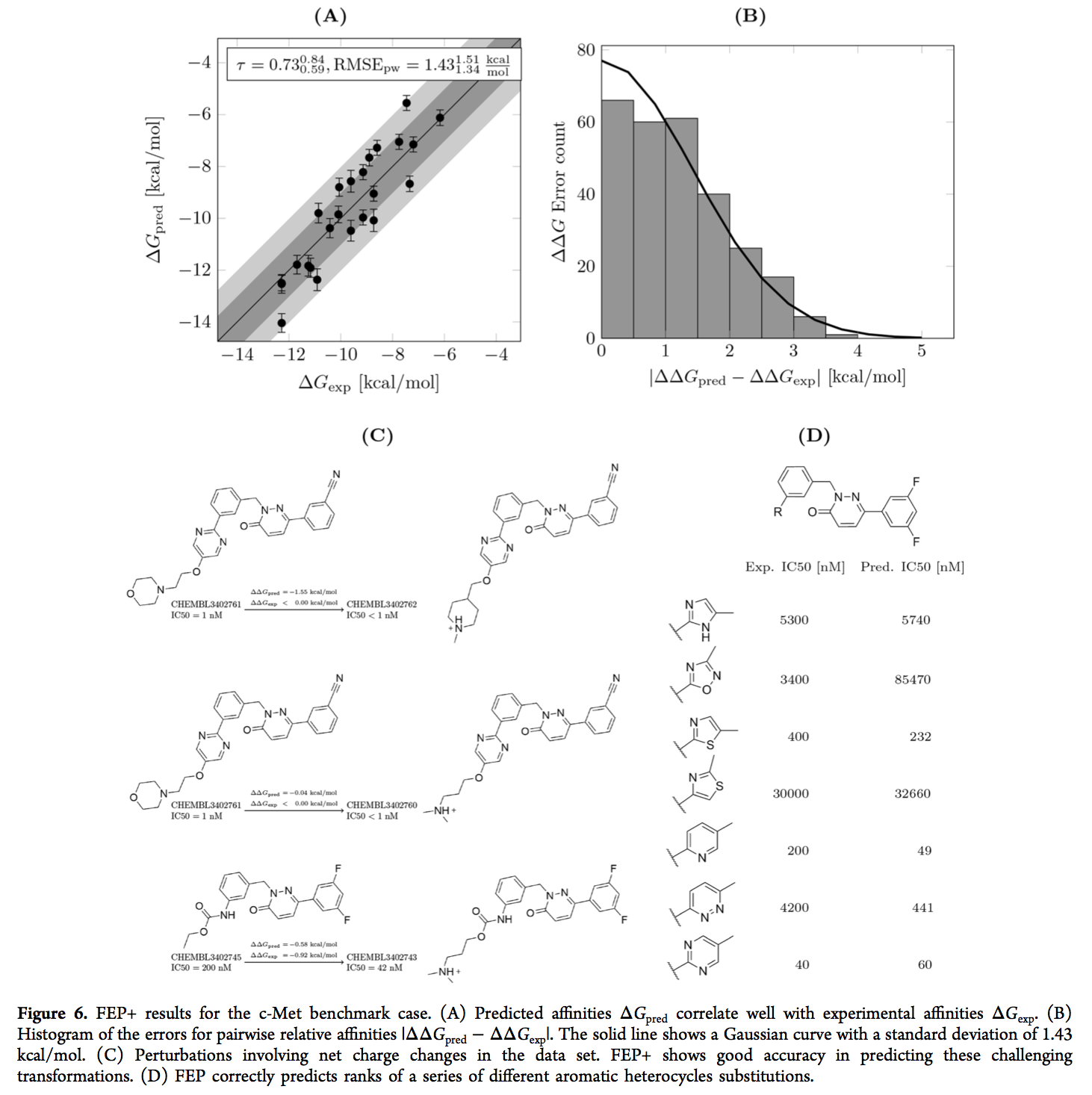
对于c-Met情况,FEP +预测重现了从氨基甲酸酯单元变为各种芳族杂环的SAR(图6(D))。它正确地将嘧啶列为比两种噻唑变体(咪唑,恶二唑和哒嗪)更有效的嘧啶。但是,当从吡啶变为嘧啶时,它没有再现效力的增加(这两种具有结合亲和力分别为IC50 = 200 nM和IC50 = 40 nM的化合物的预测分别为49 nM和60 nM )。这种变化仍在方法的典型误差范围内(1 kcal / mol)。我们最初假设吡啶化合物的质子化物种的存在会负面影响铰链结合基序,因此吡啶和嘧啶化合物质子化状态的质子化状态不同可能解释了效价的差异,但是Jaguar的pKa计算(82) )对于吡啶化合物产生的pKa为4.5,这使得带电物质的存在极不可能发生(对于嘧啶化合物的pKa为1.9)。对于吡啶化合物的预测值与实验值的观察到的偏差的另一种可能的解释可能是在结合位点缺乏杂环的旋转异构体的采样。由于其对称性,嘧啶化合物可以两种旋转状态结合,通过N原子接触蛋白质的铰链区。相反,吡啶只能以一种构象结合以建立这种相互作用。因此,如果在模拟过程中仅取样一种构象,则对嘧啶的结合具有熵上的有利贡献,而这是没有考虑的。确实,嘧啶和吡啶化合物的轨迹表明,在复杂的模拟中旋转受到阻碍,而在溶剂中,两种状态均被采样(示例显示在图S6中)。
对于SYK基准案例,我们发现精度较低(RMSEpw = 1.611.541.67 kcal / mol,请参见图7和表1)。 这导致预测的亲和力与实验亲和力之间的相关性较差(Kendallτ= 0.290.110.46)。 有趣的是,尽管化合物保留在结合位点并保持与铰链区的相互作用,但在所有模拟中,对于至少一种配体(> 3Å),配体RMSD的波动较大。 尽管如此,这种流动性可能表明采样困难。

一组离群值与化合物CHEMBL3265015相关。 我们观察到,涉及该化合物的六个扰动中有四个扰动显示绝对误差> 1 kcal / mol(一个示例在图7(C)中显示)。 化合物CHEMBL3265015在邻位的苯环上带有甲基,这可能会影响环的旋转。 与c-Met情况类似,在复合物中沿该扭转角的采样不完整(数据未显示)。
当从化合物CHEMBL3265009转换为CHEMBL3265003时,我们还注意到一个较大的异常值(|ΔΔGpred–ΔΔGexp|> 2 kcal / mol,图7(C))。在这里,分子是通过两个延伸到溶剂中的芳环生长的。类似地,根据实验,从CHEMBL3264999到CHEMBL3265003的预测相对亲和力被高估了2 kcal / mol以上。对于每个λ窗口以20 ns采样进行计算时,观察到了相同的结果。但是,准确预测了从CHEMBL3265003到大小相似的分子CHEMBL3265004的扰动(绝对误差= 0.66 kcal / mol)。我们在PFKFB3中对化合物37(参见图S4(B))以及我们在一些内部项目中的扰动也观察到了类似的高估。这种对结合袋入口处生长和置换溶剂的基团的高估可能是由于水模型高估了水的流动性或低估了水与蛋白质的相互作用。最近,几个研究小组发现,通过增加水-蛋白质相互作用强度的力场可以更好地捕获内在失调的蛋白质。(83-85)对不同水模型和水-蛋白质相互作用参数的系统研究可能是一个有趣的课题未来的研究。
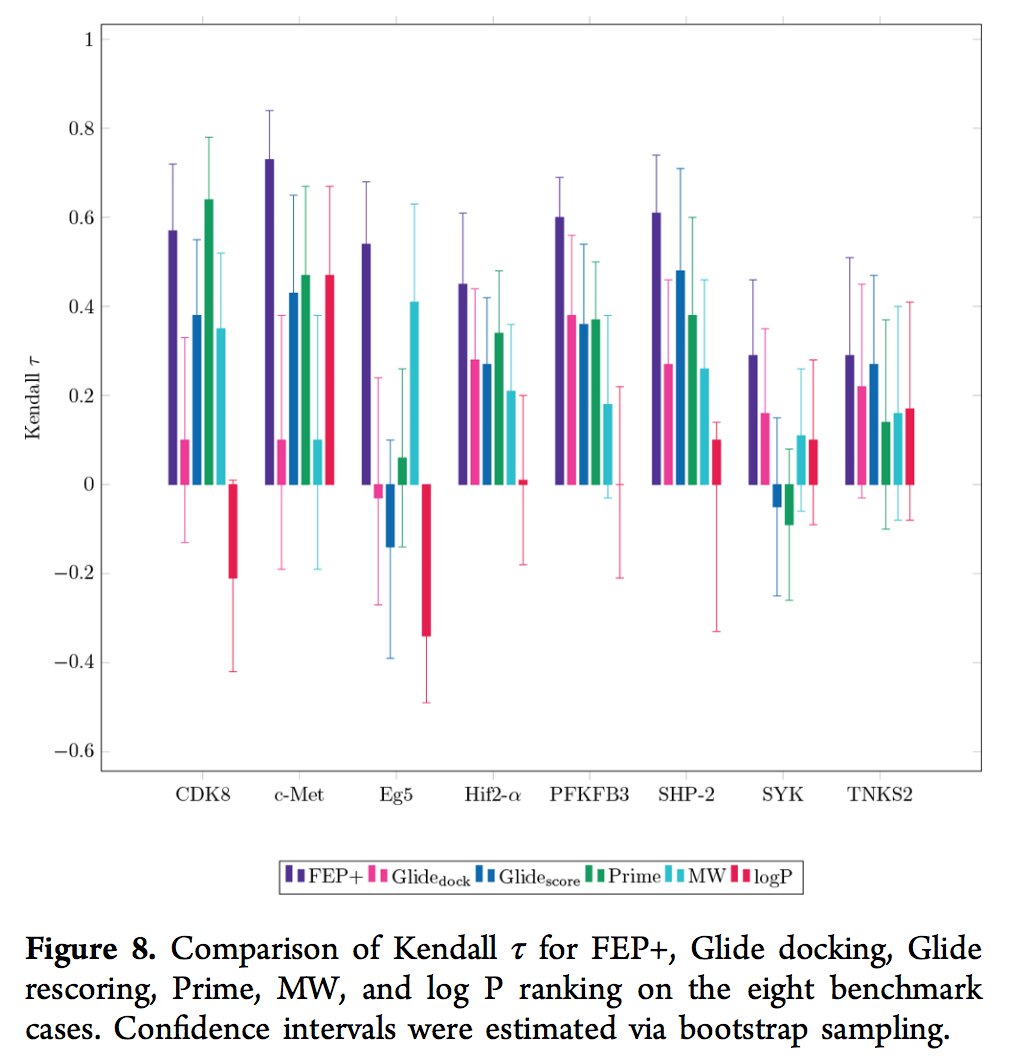
将基准上的FEP +结果与滑行对接,滑行计分和Prime MM-GBSA计算的结果进行比较,我们发现FEP +与八种情况中的七种的实验数据表现出更好或同等的相关性(表1,表S5和图5)。 8)。 在此基准上,FEP +明显胜过三种更简单的方法(与Glide对接,Glide rescoring和Prime相比,Kendallτ的Cohen d分别为1.64、1.41和1.41,表明效果尺寸非常大(69,70)) 。 此外,它在两个分子描述符上的表现优于“零模型”(图8和表S6; Kendallτ的Cohen d分别为2.38和2.00,分别与分子量和计算的log P进行比较,表明a 巨大且非常大的效果大小(69,70))。 该结果与从我们内部项目获得的数据在质量上吻合。
重要的是要指出,本研究中提出的基准结果是在行业背景下获得的,没有遵循我们的标准协议进行广泛的优化。通过更彻底的模型优化,很有可能获得更高的精度结果。确实,对于Eg5数据集,我们发现蛋白质结构-特别是配体附近的一个环-对预测的准确性有很大的影响。我们可以清楚地将此结构变化与一组与该环接触的酚基异常值联系起来(见图S7)。但是,在药物开发项目的背景下,验证和模型优化通常只有有限的时间(通常为2周)。我们认为,此处报告的性能为在行业环境中使用FEP +时可以预期达到的精度提供了现实的看法。我们希望此设置将对推动该领域的进一步方法开发有用。可以在 www.github.com/MCompChem/fep-benchmark (标记v1.0)上找到8个基准案例的输入结构以及5 ns和20 ns的FEP +结果。
2.5 FEP +对项目和运营挑战的影响
尽管获得了总体上令人鼓舞的前瞻性FEP +结果,并且与简单的SBDD方法相比具有明显的优势,但在整个计划中,我们清楚地知道,预测的有用性和达到有意义的排名所需的准确度水平在很大程度上取决于化学系列以及在优化阶段观察到的动态范围/亲和力分布。例如,对于目标3 /系列1和目标3 /系列3,尽管报告的RMSE大于1.5 kcal / mol(表S1和图3),但我们发现FEP +具有良好的排名。这有助于确定化合物的合成优先顺序,并且被项目化学家认为是有价值的。另一方面,在目标1 /系列1-3中,尽管就RMSE而言具有相似的准确性,但我们没有获得任何预测性排名。根据我们的经验,在化学系列潜在效能分布显示较大变化的项目中,可以容许较低的准确性。一旦优化达到了化学空间中的亲和力“峡谷”,其中小的变化不会对效能产生很大的影响,那么应用自由能计算对项目的价值就非常有限(就像目标1 /系列1-3和目标1的情况一样) 5 /系列1)。有趣的是,我们发现这种情况经常出现在(后期)潜在客户优化项目中。在该计划开始之初,我们认为lead优化应该是FEP的主要用例,因为通常在此阶段进行的小规模化学转化最适合该方法。然而,与我们最初的预期相反,我们注意到当化学空间得到更广泛的探索并且效价仍然是主要的优化参数时,我们对铅到铅优化或片段优化的影响更大。此外,在这一点上,化学资源通常更加有限,化学家仍然面临合成挑战,这些挑战限制了可以合成和进行实验测试的分子数量。在这种情况下,优先考虑想法以专注于最有前途的想法被认为是非常有价值的,并且执行这些计算量大的计算不被视为瓶颈。此外,指导设计团队应对应制造的化合物产生的影响要比建议他们不应制造的化合物产生的影响更大。很难轻易地评估后者对优化的影响。在适用范围方面,必须牢记,在命中最优化阶段可能会限制可预测性,因为在这些早期阶段遇到的化学转化通常对该方法更具挑战性。
强有力的沟通对于成功实施项目中的free energy计算也至关重要。我们经历了在项目团队中充分了解该方法的功能和局限性是多么重要。这有助于选择适用范围内的化合物,以FEP +优先处理。最初,我们在项目中使用FEP +时面临挑战,因为提交用于计算的想法不在该方法的范围内。在计划的后期阶段,我们专注于对定制库( libraries )进行排名,这些库在设计上更适合于计算。这种方法的一个缺点是,在某些情况下,此类库扫描的结果由项目团队使用,并用于新分子的设计;但是,这些信息与其他想法结合在一起。这样,由于未合成FEP预测的确切分子,因此很难评估该方法的影响。为避免此问题,我们旨在稍后使用FEP +预测此类分子,以便能够通过我们的自动工作流程评估预测的质量(请参见上文)。总的来说,我们发现当结果可以被解释或合理化时,化学家对FEP预测的接受程度更高。例如,通过分析相互作用或配体柔性。因此,我们建议在进行此类分析的同时提供FEP预测,尤其是关注那些被预测为最佳和最坏粘合剂(binders)的化合物。
总而言之,为了在项目中充分利用自由能计算,必须仔细权衡平衡预测准确性,适用范围,关键的优化挑战,所关注化学系列的合成可及性以及项目时机。 我们发现,筛选大型定制库(理想情况下要考虑非效能优化参数)是提供附加值的有效方法。 对于这些库,我们筛选至少50–100个想法。 我们的目标是筛选出比合成中可以选择的最大化合物数量多5到10倍的想法(这与最近的出版物相一致(86))。
三、结论
自由能计算在制药行业中越来越常用,并且已成为计算化学家工具箱中的一项强大功能。在这里,我们报告了在大量内部药物发现项目中前瞻性使用FEP +的数据。我们为各种目标和化学系列获得了良好的准确性。然而,我们能够获得高精度预测(RMSE <1 kcal / mol)的目标数量有限。这些结果与在新的公开基准测试集上获得的结果一致。该基准数据集的准备好的输入结构可供社区使用,并可能推动方法的进一步开发。在现实世界中的药物设计项目中,并不总是给出具有大亲和力扩散的配体系列的结构信息和可用性。除了预测的准确性外,我们还确定了影响该方法在项目中的影响的多个重要操作因素。在不久的将来,我们将FEP +设想为一种专家工具,以支持具有大规模库计算功能的启用FEP的项目。
四、实验部分
4.1 蛋白质结构制备 Protein Structure Preparation
蛋白质结构可从PDB( www.rcsb.org )或从我们的内部数据库下载并导入Maestro。(87)如果存在多条链,并且没有迹象表明多聚体是生物学相关形式,则结构是拆分,并且每个链都分别进行处理。使用Prime Homology模型( 88,89)去除了结晶构建体(如果有)中引入的突变,并构建了缺失的侧链和缺失的环。如果添加了循环,则随后使用Prime循环优化协议对其进行优化。序列比对使用ClustalW进行,必要时手动进行调整。使用基于知识的方法并考虑了抑制剂,建立了同源性模型。为了完善循环,使用了根据循环长度建议的设置。如果缺少较大的结构域(超过20个残基),则不添加结构的这些部分。如果有给定目标的多种结构可用,则还会根据替代结构对环结构进行建模。然后用Maestro中的蛋白质制备向导对结构进行处理。(87,90)加入氢气,保留所有结晶水,并将末端加盖。用Epik评估共晶配体的质子化状态。(91)用PropKa测定蛋白的质子化状态,(92,93)并优化氢键。最终,通过0.3Å(默认设置)的RMSD截止将结构最小化。
4.2 配体结构制备 Ligand Structure Preparation
Corina用于基于2D表示生成3D坐标。然后用LigPrep处理结构,枚举可能的立体异构体并用Epik分配质子化状态。(91,94)为了将配体定位到结合位点,我们使用了柔性配体比对工具或Glide核心约束对接(95),基于参考结构(大多数情况下是X射线配体)。核心是通过最大的通用子结构或由涉及核心变化的分子的定制SMARTS模式定义的。我们使用标准精度设置运行了Glide核心约束对接。目标1、2和3主要是通过柔性配体对准制备的。对于所有其他目标,我们最初使用这两种方法,并在整个评估过程中几乎完全切换到Glide核心约束对接。目视检查姿势,并且在多种可能的结合模式(例如,对于不对称取代的环),多种可能的互变异构体或电荷状态的情况下,结合姿势FEP用于评估所有姿势和状态。具有最低自由能的姿态/状态被用作最终输入结构。用力场生成器分析结构以检测缺失的扭转参数。如有必要,通过“力场生成器”协议生成新的自定义参数。
4.3 内部项目中的预期自由能计算
使用Schrödinger套件版本2016-4至2018-3的SchrödingerFEP +方法(5)进行了预期的自由能计算。在2018-1版之前,OPLS3力场(15)与由力场生成器生成的自定义参数一起使用。对于使用2018-2版及更高版本执行的计算,使用具有自定义参数的OPLS3e力场(17)。根据验证研究确定的最佳设置进行前瞻性计算。在大多数情况下,这些与默认设置相对应。在某些情况下,每个λ窗口的采样扩展到5 ns以上。使用FEP + GUI对计算进行了分析。(87)如果发现收敛问题,则在时间和计算资源允许的情况下扩展仿真。在所有情况下,未收敛的边和具有高滞后值的边都将从最终贴图中移除。最后,将所有大于-5.81 kcal / mol的预测ΔG值设置为-5.81 kcal / mol,以便与实验更好地进行比较(测定底部的典型值)
4.4 内部体外检测 In-House in Vitro Assays
对于除了靶标8和靶标2之外的所有靶标,使用生化酶活性测定法来确定分子的结合亲和力。 对于靶标8,将功能和ITC测量值的混合物用于与FEP +结果进行比较(在功能分析中未显示活性的分子未使用ITC进行分析)。 对于靶标2,使用细胞表型测定法确定亲和力。
4.5 基准数据集的自由能计算 Free Energy Calculations on Benchmark Data Set
完整的数据集总结在表1中。蛋白质共晶体结构可从蛋白质数据库( www.rcsb.org )下载。配体的结构和亲和力(IC50)可以从论文和专利中手动提取(71-80),也可以通过ChEMBL提取。(96,97)如上所述,蛋白质结构和配体结构可用于自由能计算。与内部数据集相比,使用X射线配体作为参考,通过Glide核心约束对接(95)排列配体结构。对于基准测试,使用Schrödinger套件版本2018-3和OPLS3e力场进行FEP +的自由能计算。(17)将准备好的结构加载到FEP +面板中,并添加亲和力数据。使用默认设置(最佳拓扑)生成地图。没有对地图进行进一步的修改。 FEP +作业在每个λ窗口中运行5 ns和20 ns的采样时间。使用Maestro中的FEP +面板分析输出。与前瞻性计算相比,我们没有修改最终图谱(例如,删除具有高滞后性的边)以提高结果的可重复性。
4.6 与其他方法的比较
为了进行比较,我们使用SchrödingerSuite 2018-3中的Glide标准精密配体对接工作流程(95)将配体对接到各自的蛋白质结构中,并使用默认设置。作为受体,使用了用于自由能计算的蛋白质结构(删除了所有水分子)。根据Glide gscore对配体进行排名,并将该得分与实验亲和力进行比较。我们将此称为“ Glidedock”方法。此外,我们仅使用Glide gscore对FEP +输入姿势进行了评分。为此,我们将Glide标准精度工作流程与配体采样“无(仅优化)”配合使用。所有其他设置保持默认值。 FEP +输入配体的姿势在受体区域内降至最低,然后评估Glide gscore得分。我们将此称为“ Glidescore”方法。使用默认设置使用Schrödinger套件2018-3中的Prime MM-GBSA模块执行MM-GBSA计算。没有水分子的蛋白质结构被用作受体,配体的姿态取自FEP +输入。根据“ MMGBSA dG Bind”得分对配体进行排名。使用Maestro计算中性配体的分子量和logP
4.7 分析
使用Maestro中的FEP + GUI进行收敛,滞后( hysteresis )和相互作用分析。 使用Python numpy,scipy(98)和自举程序库评估了相关统计信息和错误。 所有性能指标的90%对称置信区间(90%CI)使用引导程序通过对所有具有替换的数据集进行重新采样(具有10000个重新采样事件)来计算。 估计所有性能指标的配置项,并报告为xxlowxhigh,其中x是从完整数据集(例如,RMSE)计算出的平均统计量,xlow和xhigh是该统计值在已排序值的第五个和第95个百分点 引导程序样本列表。 使用内部自定义脚本对我们的内部数据库中的预期FEP计算进行数据整理和实验数据提取。 使用以下公式将IC50或Kd值转换为自由能
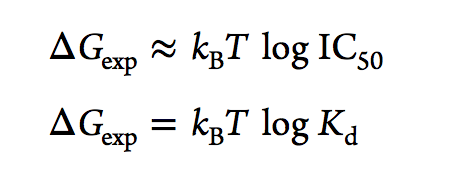
请注意,与应用Cheng-Prusoff方程相比,该近似值导致ΔG值的恒定偏移。(99)这对我们关于化合物的相对自由能排名的结果没有影响。 但是,这意味着此处给出的结果将无法直接与计算出的绝对结合自由能进行比较。
为了根据实验结合亲和力测定法的误差,估算每个数据集的最大可实现性能,(53)我们基于高斯分布生成了1,000套结合自由能,均值μ=ΔGexpi,标准偏差σ= 0.4 kcal / (5)将计算出的相关统计的结果分布的平均值,第五个百分数和第95个百分位数报告为最大可实现的性能(“最大”)。 该模型反映了两个独立实验测量值及其不确定性之间的预期相关性。
参考资料
- Large-Scale Assessment of Binding Free Energy Calculations in Active Drug Discovery Projects . https://dx.doi.org/10.1021/acs.jcim.0c00900
