【2.5.1.6】使用正态模式分析(Nmode)计算雌激素受体和雷洛昔芬复合物的熵
一、设置为普通模式计算
请注意,自2010年5月起,正常模式的计算是通过nab程序mmpbsa_py_nabnmode完成的,该程序由nab在此处描述的常规安装过程中编译。该程序必须存在于PATH或$ AMBERHOME/exe中才能运行计算。 mmpbsa_py_nabnmode和nmode之间的主要区别在于nab程序可以在Generalized Born溶剂中计算正常模式,因此又添加了两个输入变量(请参见手册中的nmode_igb和nmode_istrng)。此实现还应该改善在脚本的早期版本中看到的最小化收敛问题。
二、建立起始结构并运行模拟以获得平衡的系统
我们将在此模拟中建模的系统是雌激素受体蛋白质和雷洛昔芬配体之间的蛋白质-配体复合物。这是该组合系统的预先准备的pdb文件。
Estrogen_Receptor-Raloxifene.pdb
该结构包含雌激素受体蛋白以及称为Raloxifene的配体,该配体先前已与该蛋白对接,如下图所示:
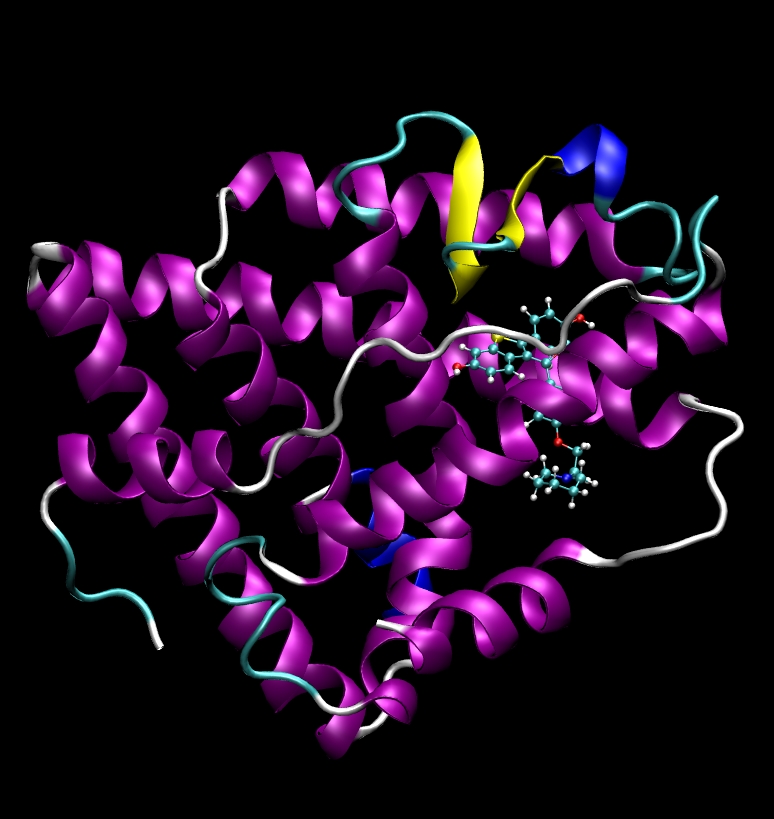
该系统的构建方式与第1节中的Ras-Raf系统类似。有关如何构建起始结构和运行模拟以获得平衡系统的说明,请参阅第1节和第2节。请注意,您还必须使用antechamber获取雷洛昔芬的正确参数。有关详细说明,请参见Sustiva教程(基本4)。
MD模拟中使用MMPBSA.py计算结合自由能的重要文件是拓扑文件和mdcrd文件(Est_Rec_top_mdcrd.tgz)
三、使用普通模式分析(普通模式)计算绑定熵
现在,我们将计算复合物,受体和配体的正常模式,并对结果取平均值,以获得结合熵的估计值。请注意,可以通过取消注释常规部分的最后一行以使用AmberTools中的ptraj程序执行准谐波熵计算来计算系统的熵贡献。
我们将使用以下针对MMPBSA.py的输入文件执行正常模式计算:
mmpbsa.in
Input file for running entropy calculations using NMode
&general
endframe=50, keep_files=2,
/
&nmode
nmstartframe=1, nmendframe=50,
nminterval=5, nmode_igb=1, nmode_istrng=0.1,
/
MMPBSA.py的输入文件被设计为类似于AMBER的sander模块中使用的mdin文件的设置。每个部分的开头均以“&”号表示,后跟该部分的名称。此外,反斜杠(/)或'&end’可用于结束该部分。有关所有变量的完整列表,请参见amber手册。该输入文件分为两个名称列表:&general和&nmode。 &general名称列表指定的变量不是特定于任何特定计算,而是特定于所有计算。在这种设置中,雌激素受体是受体,雷洛昔芬是配体。 ‘endframe’变量设置使用ptraj制作干mdcrd文件时停止在mdcrd的哪一帧。 “ keep_files”变量允许用户指定在计算结束时删除哪些文件。 nmode部分用于定义特定于正常模式计算的变量。 ‘nmstartframe’变量定义了从正常模式分析开始的干mdcrd开始的帧。 ‘nmendframe’和’nminterval’分别设置干帧的正常模式分析的结束帧和间隔。请注意,nmstartframe / endframe / interval变量对应于由&general中定义的开始帧,结束帧和间隔提取的快照的“轨迹”。因此,将仅选择这些帧的子集用于正常模式计算(最多,_MMPBSA_complex.mdcrd中的每个帧)
$AMBERHOME/bin/MMPBSA.py -O -i mmpbsa.in -o FINAL_RESULTS_MMPBSA.dat -sp 1err.solvated.prmtop -cp complex.prmtop -rp receptor.prmtop -lp ligand.prmtop -y *.mdcrd > progress.log
或通过将脚本提交到排队系统(例如PBS)
qsub nmode.job
nmode.job
#!/bin/sh
#PBS -N nmode
#PBS -o nmode.out
#PBS -e nmode.err
#PBS -m abe
#PBS -M email@domain.edu
#PBS -q brute
#PBS -l nodes=1:surg:ppn=3
#PBS -l pmem=1450mb
cd $PBS_O_WORKDIR
mpirun -np 3 MMPBSA.py.MPI -O -i mmpbsa_nm.in -o FINAL_RESULTS_MMPBSA.dat -sp 1err.solvated.prmtop -cp complex.prmtop -rp receptor.prmtop -lp ligand.prmtop -y 1err_prod.mdcrd > progress.log
这会将计算进度打印到文件progress_nm.log中。 计算过程中的所有错误也将被打印到该文件中。 最后,一旦计算完成,将打印计时,显示脚本每个步骤所花费的时间。 如果以串行方式运行,则在10帧(frames)上运行此计算大约需要40个小时。 请注意,MMPBSA.py.MPI可用于并行运行(有关运行此程序的详细信息,请参见第3.4节)。 如第3.4节所述,这将在启动的线程数之间平均分配帧数。 系统的大小会显着影响计算时间和所需的内存(RAM)。 Segmentation faults (segfaults) are usually caused by insufficient RAM in your system.
progress_nm.log
ptraj found! Using /share/local/lib/amber10/i686/bin/ptraj
sander found! Using /share/local/lib/amber10/i686/bin/sander (serial only!)
nmode found! Using /share/local/lib/amber10/i686/bin/nmode
Preparing trajectories with ptraj...
50 frames were read in and processed by ptraj for use in calculation.
Starting sander calls
Starting nmode calculations...
Timing:
Processing Trajectories With Ptraj: 0.240 min.
Total Harmonic nmode Calculation Time: 2363.906 min.
Output File Writing Time: 0.018 min.
Total Time Taken: 2364.165 min.
MMPBSA Finished. Thank you for using. Please send any bugs/suggestions/comments to mmpbsa.amber@gmail.com
命令行上的’-y * .mdcrd’告诉脚本读取工作目录中所有以’.mdcrd’结尾的文件,并将它们用作要分析的轨迹。
将’keep_files’设置为2,以下是所有输出文件:Nmode_output.tgz。
该脚本使用ptraj创建了三个非溶剂化的mdcrd文件(复合物,受体和配体),这是在熵计算过程中分析的坐标。 使用每个快照的PDB文件执行正常模式计算,因此将从干燥的mdcrd文件中为复合物,受体和配体创建10个PDB文件。 如果熵== 1,则将平均pdb文件创建为平均结构,以对齐轨迹,以进行ptraj执行的准谐波熵计算。MMPBSA.py创建的所有文件均应以前缀’MMPBSA‘开头,但最终输出除外 文件:FINAL_RESULTS_MMPBSA.dat
FINAL_RESULTS_MMPBSA.dat
| Run on Thu Feb 11 12:44:26 EST 2010
|Input file:
|--------------------------------------------------------------
|Input file for running entropy calculations using NMode
|&general
| endframe=50, keep_files=2,
|/
|&nmode
| nmstartframe=1, nmendframe=50,
| nminterval=5,
|/
|--------------------------------------------------------------
|Solvated complex topology file: 1err.solvated.prmtop
|Complex topology file: complex.prmtop
|Receptor topology file: receptor.prmtop
|Ligand topology file: ligand.prmtop
|Initial mdcrd(s): 1err_prod.mdcrd
|
|Best guess for receptor mask: ":1-240"
|Best guess for ligand mask: ":241"
|Ligand residue name is "RAL"
|
|Calculations performed using 50 frames.
|Poisson Boltzmann calculations performed using internal PBSA solver in sander.
|
|All units are reported in kcal/mole.
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
ENTROPY RESULTS (HARMONIC APPROXIMATION) CALCULATED WITH NMODE:
Complex:
Entropy Term Average Std. Dev.
-----------------------------------------------------------
Translational: 16.9389 0.0000
Rotational: 17.3953 0.0038
Vibrational: 2784.7967 2.3982
Total: 2819.1307 2.3964
Receptor:
Entropy Term Average Std. Dev.
-----------------------------------------------------------
Translational: 16.9233 0.0000
Rotational: 17.3911 0.0045
Vibrational: 2755.5693 3.0352
Total: 2789.8840 3.0342
Ligand:
Entropy Term Average Std. Dev.
-----------------------------------------------------------
Translational: 13.2972 0.0000
Rotational: 11.4991 0.0496
Vibrational: 33.7549 0.0442
Total: 58.5511 0.0058
DELTA S total= -30.0192 +/- 3.4064
NOTE: All entropy results have units kcal/mol. (Temperature has already been multiplied in as 300. K)
-------------------------------------------------------------------------------
-------------------
输出文件的开头包括有关计算的各种详细信息。 输出文件的其余部分包括每个平移,旋转,振动和总熵贡献的平均值,标准差和均值的标准误差。 在这些部分之后,将给出ΔS以及标准偏差和均值std误差。
人们通常会期望找到生物复合物的负ΔS值。 这象征着当蛋白质和配体结合形成复合物时,可用的微状态减少。 可用微状态的减少主要是由于配体被捕获并在与蛋白质结合时具有有限的迁移率。
从负ΔS值-30.02 kcal / mol,我们可以清楚地看到这是纯水中的一种熵不利的蛋白质-配体复合物,但请记住,由于我们没有估算焓对绑定的贡献(有利),因此该结果不等于真实的结合自由能。
参考资料
