【4.7.3.1】均方根差(RMSD)
一、基本概念
RMSD 是最常用、最经久不衰的蛋白结构度量,也最被诟病。它定义简单,就是两个结构对应原子坐标差值的均方根(公式1)。
定义分子的“惯性半径”(Rg,radius of gyration)如公式2,那么,RMSD 的公式可以由 Rg 表达(公式3)。这里注意,两个结构以最佳方式重叠后,质心应当重叠,因此可以不失一般性地令其质心在原点,在公式2中约去向量 rc。[4] 因为计算 RMSD 必须先将相比较的2个结构最大化地重叠,即找到一个刚体变换,令 RMSD 最小,所以 RMSD 的计算公式可以更数学化地写为公式4(考虑原子质量 mi)。
AlphaFold2 用的是 RMSD95,即只考虑95%的残基最大化的重叠(公式5),这样降低了一些灵活区域的影响。[1]
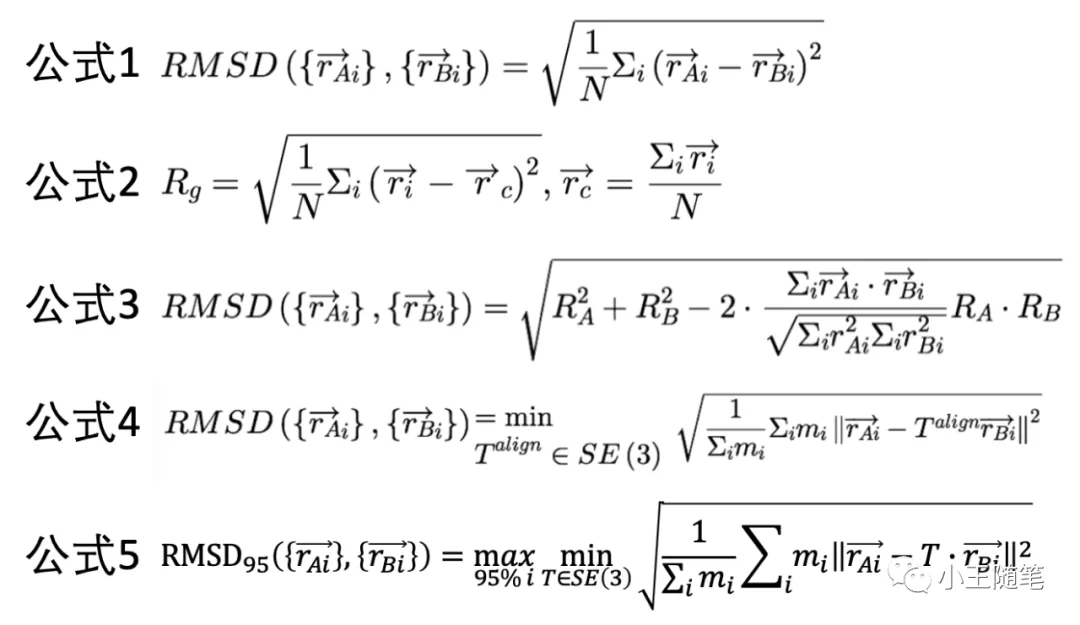
虽然 RMSD 计算简单,物理意义明了,但它有一些明显的缺点:
- 计算强烈依赖于两个结构的重叠对齐算法;
- 无法区分整体的结构拓扑;
- 对结构的缺失部分不敏感;
- 有链长依赖。
对2、3,通常,边角灵活区域的结构浮动所造成的 RMSD 差异,可能盖过整体结构的重叠。也就是说,即便2个结构整体相似(有相同的fold),其二者也会因为少部分灵活区域的不同而有较大的 RMSD。
对4,同样的 RMSD 值对较小的蛋白可能意味着巨大的结构差异,对较大的蛋白则可能代表微小的结构差异。这一点我们可以由公式4看出—— Rg 显然依赖于蛋白的大小。实际上,
为了回应 RMSD 的上述缺点,诸多 RMSD 变体被设计出来,我们介绍2个:rRMSD和Z-rRMSD。
2001年,Skolnick 与学生 Betancourt 设计出 relative-RMSD,即 rRMSD [4],来消除链长依赖 —— 着眼点就是公式3。
考虑“对齐相关系数”(aligned correlation coefficient, ACC),定义一组随机结构的 ACC 的平均值为 C(N)(公式6)。在考察了1300个同源性低于30%的结构的连续残基片段后,作者获得了 C(N) 的一个估值(公式7)。—— 21年后的今天看,这个公式是这篇文章最有价值的成果,而不是它的 rRMSD。
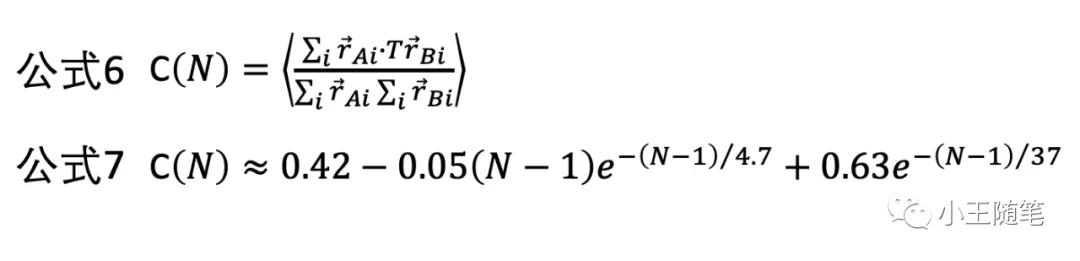
公式7的两个特征幂指数 4.7 和 37 的单位是“残基”。
4.7 个残基大约对应 2.8 nm,与球蛋白的 Lp 值接近(persistence length)。Lp 是高聚分子的分子链灵活性的特征值。它表示一条高分子链上的两点,在相距一定距离后,其各自的取向彼此不相关(decorrelated)。设想一手拽住一根长绳抖动,绳上相距很近的位置的取向必然相互关联,换句话说,某一处的位置和运动速度,将在一定距离内,影响它后面部分的位置和运动;但是,当两点相距足够远,这种相关性就没有了。
37 个残基,作者认为,是其所考察的中等长度多肽链(片段)的相关长度。虽然有强行解释的味道,但从物理含义的角度理解公式依然很有意思。
有了C(N),可以估计随机的两个多肽结构可能有的平均 RMSD(公式8),进而定义 rRMSD(公式9),这样就消除了链长依赖,改进了 RMSD 的缺点4。
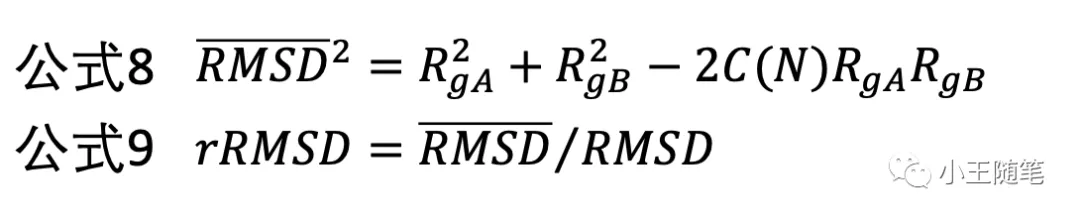
一脉相承地,2004年,Skolnick 与学生张阳基于 rRMSD,设计了 Z-rRMSD(公式10,公式11),一种类 Z-score 的统计重要性(statistical significance)的度量。[5]
Z-rRMSD 从概率上判断2个结构(片段)是否相似,更重要地,是否基于已知所有结构”universally”相似。如果2个结构的 Z-rRMSD 打分很高,那么这2个结构很可能有相同的 fold。也就是说,Z-rRMSD 改进了RMSD 的缺点2、3。
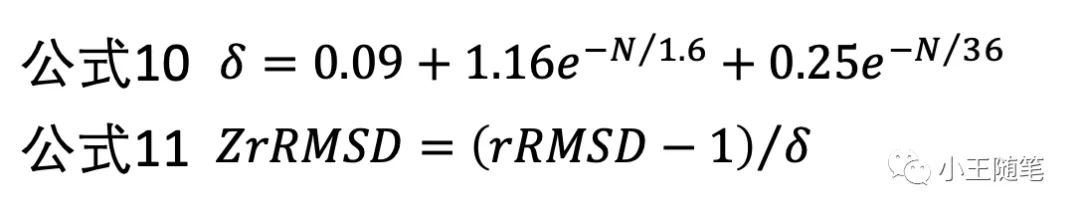
”有相同的 fold“?这不是 TM-score 所擅长的吗?对。文献5就是提出 TM-score 的著名论文。Z-rRMSD 可以作为 TM-score 的替代。
三、工具
RMSD: Root Mean Square Deviation 是一种在分子模拟及预测中很常见的评价标准,通过Jacobi变换来的到一个大分子和目标分子的相似程度。常用来评价一个三维结构的预测结果是否足够准确.
有时我们需要计算两个(或更多)分子之间的RMSD,可以用来判断两个构象的接近程度。
好用的工具:
https://github.com/charnley/rmsd
用法:
pip install rmsd
import rmsd
import numpy as np
P = np.array([[-0.9835 , 1.8109 , -0.0314 ],
[ 0.1268 , 1.8041 , -0.03242],
[-1.4899 , 3.2274 , 0.18102],
[-1.3504 , 1.1535 , 0.78475]])
Q = np.array([[-2.1217 , 4.0933 , 0.12713],
[-1.0113 , 4.0865 , 0.12611],
[-2.628 , 5.5097 , 0.33955],
[-2.4885 , 3.4358 , 0.94328]])
print "RMSD before translation: ", rmsd.kabsch_rmsd(P, Q)
P -= rmsd.centroid(P)
Q -= rmsd.centroid(Q)
print "RMSD after translation: ", rmsd.kabsch_rmsd(P, Q)
参考资料
- https://github.com/charnley/rmsd
- https://mp.weixin.qq.com/s/6KGsazYa5MXtCTkwaogBjA
- [4] Betancourt & Skolnick, Universal similarity measure for comparing protein structures, Biopolymers (2001)
- [5] Zhang & Skolnick, Scoring function for automated assessment of protein structure template quality, Proteins (2004)
