【4.7.1.3】三级结构的比对
结构比对就是对蛋白质三维空间结构的相似性进行比较。他是蛋白质结构分析的重要手段之一。
- 可用于探索蛋白质进化以及同源关系
- 改进序列比对的精度
- 改进蛋白质结构预测工具
- 为蛋白质结构分类提供依据
- 帮助了解蛋白质功能
结构比对的结果可以用很多种参数来衡量,最常用的是 root mean squared deviations(RMSD)。如果两个结构的RMSD为0埃,那么他们结构一直,可以完全重合;一般来说,RMSD小于3埃时,认为两个结构相似。
三维结构的相似性比较可归纳为三大类:
- 基于空间特征分布的比较
- 基于几何的比较
- 基于拓扑的比较
比较重要的蛋白质两两结构比较的算法包括:CE, DALI, SSAP, Geometric Hshing ,MatAlign
目前,人类对蛋白质的认识还只是冰山一角。对基因所表达的各种蛋白质都从实验上进行研究是不切实际的, 新测得的蛋白质可通过与已知特性的蛋白质进行结构或序列比较后推知其生物功能。在生物的演化过程中,蛋白质的结构比其序列更保守[13] [14], 序列变化不一定会改变蛋白质的结构, 相似结构的蛋白质却可能具有不同的序列[15], 而具有相似结构的蛋白质往往具有相似的功能, 因此结构比较更受重视。在活性细胞中, 蛋白质通过与其它分子的适当结合, 执行了几乎全部 的主要功能, 蛋白质的结构对其功能具有重要的意义
二、方法原理概述
2.1 蛋白质两两结构比对:
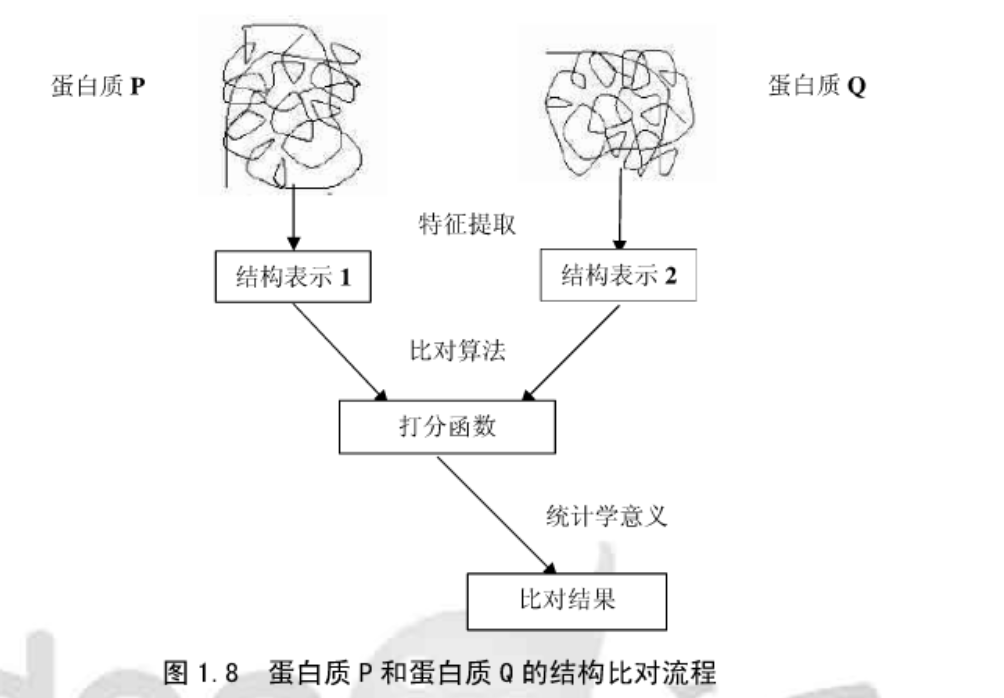
两个蛋白质结构P和Q之间的结构比对,被定义为P和Q原子之间的一一映射。通过这种映射,两个结构可以叠加并且计算二者之间的相似程度。两两结构比对的目的是为了找出给定的两个蛋白质结构的原子之间最好映射方法
- 结构表示:为蛋白质结构选择一种计算的表示方法。蛋白质有许多特性包括残基的种类,原子的位置,键的种类及特性。为了结构比对而选择的蛋白质表示方法不必考虑所有特性。许多算法只考虑一些原子的空间位置而忽略它们的种类。更简单的表示方法只需考虑主要碳原子(Cα)和Cβ)的位置。它们可用来代表残基。这样就可将一个蛋白质中需考虑的原子数目由上千个减少至几百个。
- 特征提取:基于不同的结构表示进行特征提取。大多数的特征都来自于二级结构元件和内部残基(主要指Cα或Cβ之间)的距离矩阵。
- 结构比较和比对优化: 使用提取的特征计算最初的比对。取决于优化的标准, 优化进行越好比对提高得越多。
- 有意义的评估:评估比对结果(通常指估计随机得到一个相似比对的似然值)。
2.2 基于动态规划算法的蛋白质两两结构比对
在两两蛋白质结构比较方面已经提出了许多方法[19]。它们使用不同的蛋白质结构表示方法(如用原子,残基或者二级结构元件及其组合来表示)和算法(如动态规划,几何散列法以及随机算法)。下面我们将分别对这些比对方法进行介绍。
动态规划(DP)算法已经成功应用在序列的两两比对问题上[20]。然而在结构比对中不能直接使用传统DP算法。主要是因为在结构比对中,两个残基之间的距离还要依赖于其它残基的比对。
Sali和Blundell[21试图通过旋转和变换与打分函数无关的独立特征来克服上述难点。主要使用模拟退火方法,首先通过一组氨基酸残基的属性(序列一致性,疏水性, 电荷,体积,扭角等)找到两个蛋白质可能对等的部分,然后根据残基特性的相似性构建一个二维相似矩阵。最后,采应用动态规划对两个蛋白质进行比对。
Orengo和Taylor[22]提出使用双层动态规划的SSAP方法。在外层动态规划中,得 分矩阵项Sij代表第一个蛋白质第i个残基与第二个蛋白质中第j个残基比对的得分,这样矩阵的最佳路径就是两个蛋白质结构的最佳比对,而其中ij值是由内层动态规划 计算出来的: 假定第i个与j个残基是匹配的,另一个打分矩阵是通过计算残基的Cα-Cβ 向量之间区别。这样最终的得分Sij就表示当第i个与j个残基匹配时,剩余残 基的比对情况。Taylor[23]通过加入随机步骤扩展了此算法。这种方法允许次优比对存 在,并利用双层动态规划迭代的评估次优化比对的效果。
Gerstein和Levit[24]提出了另一种迭代的动态规划方法。先随机给出一个结构比对,在此基础上,通过叠加蛋白质优化RMSD,并且计算内部残基距离;再对内部残基的距离矩阵应用动态规划方法找到最佳匹配;然后调整当前比对。重复上述过程直 至收敛。整个计算过程是使用不同初始比对,最终将输出最佳比对。
在这部分,这类算法用二维的距离矩阵来表示蛋白质结构,即由蛋白质中每个残基与其它残基之间距离构成矩阵。DALI就是其中使用比较广泛的算法,通过两个蛋白质的距离矩阵来查找具有高度相似的局部结构。算法的初步假定是如果两个蛋白质结构相似那么它们的距离矩阵也应该相似。第一步,比较两个蛋白质的距离矩阵找到大小为6个残基的相似子矩阵,这样每个蛋白质结构都被表示成六缩氨酸的组合体,极大减少了计算量,又不失比对精度,因为一般的二级结构单元分子数均大于6个氨基酸。然后将相邻的比对合并,称之为seeds。这些seeds随机合并或通过Monte Carlo算法找出最优解或次优解再继续扩展。通过随机移动部分比对和重新比对蛋白质可能得到更好比对。算法使用两种不同的得分方法:严格相似性方法和宽松的得分方法。DALI的变形通过增加二级结构元件约束条件以提高计算性能。
CE与DALI相似,使用距离矩阵找到短但高度相似的片段,两者的区别在于CE对这些片段进行组合扩展。在第一阶段,两个蛋白质中所有8个残基比对的组合都要被测试,大于阈值的选择出来。比对是由一个初始片段通过不断添加新片段而形成。如果一个新添加的片段使比对获得更高得分值,且超过某个阈值,那么就添加这个片 段。为了提高性能,算法不允许连续空位个数超过30。在找到较好比对后,算法通过再利用动态规划调整比对。
Chew[25]用邻接的Cα原子向量来表示蛋白质结构,这些向量描述了蛋白质骨架结构,并将这些向量放在单位球上并通过它们在球上轨迹来比较两蛋白质结构。
2.3 基于几何哈希法的蛋白质两两结构比对
几何哈希法是利用对几何不变性的原理对三维物体旋转和变换来进行比较。Nussinov和Wolfson[26]就是利用这种方法进行蛋白质结构的比较。首先对每种蛋白质给出一系列的参考架构。并且把蛋白质的残基映射到相应的参考架构的三维网格上。如果两个蛋白质结构相似,就会得到有大量的残基对映射到相同三维网格中的两个参考架构(每个蛋白质对应一个架构)。为了进行有效的查询,对三维网格构建立哈希函数,并且既可以用每个氨基酸残基的Cα,Cβ和N原子来定义参考架构[27],也可以一次用三个或更多的残基来定义。
Verbitsky[28]在使用几何哈希法进行结构比对时可以包含铰链弯曲结构,而Holm和Sander[29]使用几何哈希法处理二级结构元件,即参考架构对应二级结构元件对,而其余的二级结构元件则是散列的,通过查找相同位置上映射着相对应的二级结构元件的一对架构,而匹配架构在迭代优化初始比对中形成的,从而实现与参考架构对应的两 蛋白质结构的比对。
2.4 基于层次算法的蛋白质两两结构比对
层次算法是基于快速识别两个蛋白质上的相似的二级结构元件片段,这里两个片断的相似性是用长度和角的约束条件来定义的。匹配较好的片断对就构成了原子级比对的seed。
VAST程序[30使用二级结构元件进行层次比对,首先利用二分图,二分图一边的顶点表示查询蛋白质的二级结构元件。二分图另一边的顶点则表示目标蛋白质上的二级结构元件。如果两个二级结构元件匹配,就在两个顶点间建立一条边。初始的二级 结构元件比对就构成了二分图的最大完备子图,再用吉布斯采样法将这个初始的比对扩展到Cα原子。VAST程序的优点之一就是能报告通过反映的意外匹配,这里p-value是通过考虑匹配的大小、蛋白质大小,以及比对的质量等因素计算得出的。
还有其它很多的算法使用了层次比对[31][32] 。LOCK通过向量表示二级结构元件,并且通过向量间角度和距离的相似度来对二级结构元件进行比对,再利用迭代的动态规划对二级结构元件比对进行扩展。在进行蛋白质比对时,通过不断计算每个残基的最近邻残基的比对情况扩展比对数目重复这一过程直至收敛。
2.5 蛋白质多重结构比对
尽管在两两结构比对问题上已有很多算法,但是只有在多重结构比对方面提出的算法却为数不多。一个通用的方法是以两两结构比对,并在此基础上构造多重比对算法[33][34][35]。
基于星形比对方法的构建过程如下。首先,对任意的两个蛋白质都进行两两结构比对,从中选取一个和其它的蛋白质比对时具有最小的RSMD值的轴心蛋白质,然后将其他蛋白质都与这个轴心蛋白质进行比对,这样就构造了一个多重结构比对。这种方法的一个缺点是如果一次只考虑两个蛋白质则可能丢失全局模式。这可以使人想起发生在多序列比对上的问题[36]。最新的一种算法通过提取蛋白质结构中的共同子结构 来捕捉全局关系,并利用这些共同子结构比对构造的多重结构比对[37][38][39]。
Gerstein 和Levitt通过扩展迭代的动态规划进行多重结构比对。首先,对任意的两个蛋白质都进行两两结构比对,从中选取一个和其它的蛋白质比对时具有最小平均距离的轴心蛋白质,然后将其他蛋白质都与这个轴心蛋白质进行比对,这样就构造了一个多重结构比对。就是说,如果中间结构中一个位置i与第一个结构的位置j匹配, 并且与第二个结构的位置k匹配,那么位置j和位置k也互相匹配。
Guda通过扩展CE[40]来实现多重结构比对。首先计算蛋白质的两两比对并选择一 个轴心结构。然后通过轴心结构找到一个最初比对,再利用蒙特卡罗算法进行优化。
Leibovitz用几何哈希法寻找共同子结构并以此作为多重比对种子,再通过合并这些种子得到更大的种子,同时得到具有最高比对得分比对种子。Shatsky提出了一个更快捷而且不需要所有蛋白质参与的比对方法。通过把每个蛋白质作为轴心结构来获得最佳多重比对。Dior通过用二级结构元件表示蛋白质来计算公共核心结构的改进 算法进行多重结构比对。结果表明用二级结构元件代替单个氨基酸表示极大提高了比对效果。
三、 工具

- SuperPose
输入A,B两个模型,输出结果中B没有移动,只有A移动了位置。所以只需要下载移动后的A,再与B同时用VMD打开,就可以看到叠合后的结构。
- SPDBV
是一款蛋白质结构分析软件,也是一个蛋白质同源建模平台。其结构叠合功能十分出色。可进行整体智能叠合,或者选择性叠合。下载后无需安装,直接运行。
参考资料:
- 山东大学 生物信息学课题组荣誉出品 http://www.crc.sdu.edu.cn/bioinfo 巩晶老师课件
- 郎美娜 硕士论文《蛋白质三级结构比对方法及其应用》
