转录组介绍
转录组(transcriptome),额定类型细胞中全体转录本(transcript)的集合,是细胞特定时刻基因表达谱的一个快照(snapshot of expression profile)。在转录组中,既包括编码蛋白的信使RNA(mRNA),也包括不编码蛋白的mirRNA,long non-coding RNA(lncRNA)等非编码RNA。这些RNA转录本彼此协同作用,共同来调控细胞的生长,发育,凋亡等一系列重要的生理过程。因此,对于转录本的研究通常包括定性和定量两个方面。
Real-Time qRT-PCR通过对经典PCR扩增反应中每一个循环产物荧光信号的实时检测,我们可以实现对其实模板的定量分析。通过正确设定引物(primer)和探针(probe),qRT-PCR技术可以很大范围内定量的检测目标转录本的拷贝数,也即表达水平。因此长被作为转录组分析中的金标准(Gold Standard).qRT-PCR只能测定一个转录本的表达水平,同时也需要知道待检测转录本的序列,难以用来发现未知的转录本。
Microarray在高通量测序之前是主要的高通量转录本表达分析技术。微阵列(microarray),也称基因芯片(gene chip),通过将几十万个不等的探针(probe)分子固定在约1cm见方的固体片基上制成的。利用核苷酸分子在形成双链时碱基互补配对原理,microarray可以一次性检测出样本中所有与探针互补的核苷酸片段,从而快速得到样本中基因的表达谱(expression profile),因此,microarray从上世纪90年代问世以来,在生物,医学,农学等领域快速获得了广泛应用。与qRT-PCR相比,micoarray虽然在通量上有了显著的提高,但仍然需要实现确定待测转录本的序列。
EST(表达序列标签)技术通过对一个随机选择的cDNA克农进行单次测序来获得cDNA的部分序列。与microarray不同,EST是基于测序的,并不需要事先知道待检测转录本的序列。可以被用来发现新的转录本。早在1991年,当时还在NIH的Craig Venter等就开始利用EST来寻找人类的新基因。然而,由于当时测序技术通量的限制,一次EST通常只能得到几千个转录本的序列,远远无法进行全转录本水平的profiling.
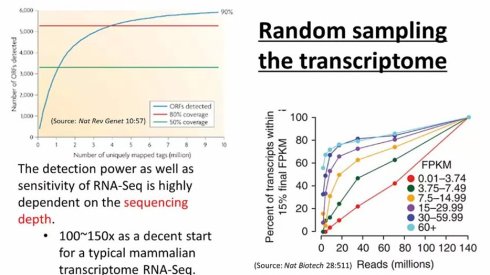
RNA-seq深度测序技术的出现,使得研究人员首次可以,在全转录组水平利用测序技术同时进行定量与定性的分析。首先,对生物样品中的RNA反转录为cDNA而后将这些cDNA打碎成较小片段后,上机测序。一方面,RNA-seq技术使得研究人员可以快速确定转录本,进而鉴定存在的可变剪切体(Alternative splicing isoform),这是传统的microarray等技术很难做到的。另一方面,对基因组特定位点上reads深度的计算,可以对表达量水平进行估计。所以,RNA-seq技术使得研究人员可以同时对转录组进行定性和定量的研究。需要注意的是,RNA-seq本质上是对转录本序列的随机抽样(random sampling),因此,其检测效力(power)和灵敏度(sensitivity)高度以来于测序深度。如果测序深度不够,就难以检测出低拷贝的基因。原则上,只有在饱和曲线(saturation curve)达到平台期(plateau)后,才能认为深度足够。对于哺乳动物转录组来说,一个经验规则是通常要做到100-150X的coverage
在随机抽样的情况下(random sampling)情况下,map到转录本上的read数目正比于其表达量(transcript abundance),因此,我们可以利用落在某个转录本上reads的总数目来估计其表达量。但另一方面,落在一个转录本上reads的书面,也于其长度和总测序深度成正比。例如有A,B两个基因,假定他们表达量相同,都转录2个转录本,但是A的长度是B的两倍,那么map到A的热啊但是数目就是map到B的reads数目的两倍。如果我们只是看这些reads的数目,我们会认为A的表达量是B的两倍,但这显然是不对的。

通量,测序深度。
所以,我们在实际分析中,通常会将原始的reads数目(raw reads count)利用线性放缩(scaling),转换为RPKM值来进行归一化(normalization)处理。

RPKM就是一个常用的归一化的方法。 这个公式里面的C是贴到这段转录本上reads的总数目,N是这次试验总reads数目(也就是测序深度),L是这段学列的长度。在假定不同样本中RNA总体分布一致的前提下,RPKM就可以正确处理由于转录本长度和测序深度引起的artifact,从而使得来自不同基因,不同sequencing run乃至不同样本之间的表达数据彼此之间可以比较。需要注意的是,RPKM并不是唯一的归一化方法。通过考虑不同的误差因素(bias effectors),引入不同的生物学假设,可以构造不同的归一化方法。事实上,已有研究表明,相比于后续提出的TMM,DESeq等方法,RPKM方法在样本差异基因表达检验等分析中的效果不是最理想。另一个需要在RNA-Seq技术中引起注意的地方是链特异性(strand-specific)。我们知道,DNA的两条链都可以转录,形成不同的转录本,然而,常用的Illumina RNA-Seq kit是不分链的,也就是说,我们无法知道配对的reads哪个方向和转录本是一致的,那个和转录本方向互补。对于分链的数据,又有两种不同的情况。在利用dUTP技术进行标记(labeling)的方法–也就是illumina的strand-specific kit 使用的方法中,第二个read和转录本方向一致,的一个read和转录本反向互补。在另一种SOLID等平台常用的secondstrand分链方法中,就刚好反过来了。因此在分析之前,我们一定要弄清楚自己的数据有没有分链,是怎样分链的。
参考资料:
此博文内容来自高歌老师的讲课
