细胞亚群鉴定--SingleR
说明文档:
近年来,单细胞RNA-seq (scRNA-seq)的研究进展使疾病模型中描述基因表达变化(gene expression )的精度达到了前所未有的水平。目前已发展出多种单细胞分析方法来检测基因表达的变化,并通过基因表达的相似性来聚类细胞。然而,根据细胞聚类进行分类在很大程度上依赖于已知的标记基因( marker genes),通常分类工作手工完成的。这种策略具有主观性,限制了密切相关的细胞亚群的分化。
本文提出了一种新的scrna -seq无偏差细胞类型识别的计算方法:SingleR(Single -cell Recognition of cell types)。SingleR利用纯细胞类型的参考转录组数据集来独立推断每个单细胞的细胞可能类型。SingleR的注释与Seurat(一个为scRNA-seq设计的处理和分析包)相结合,为研究scRNA-seq数据提供了一个强大的工具。
一、简介
1.2 原理
Step 1: Spearman correlations
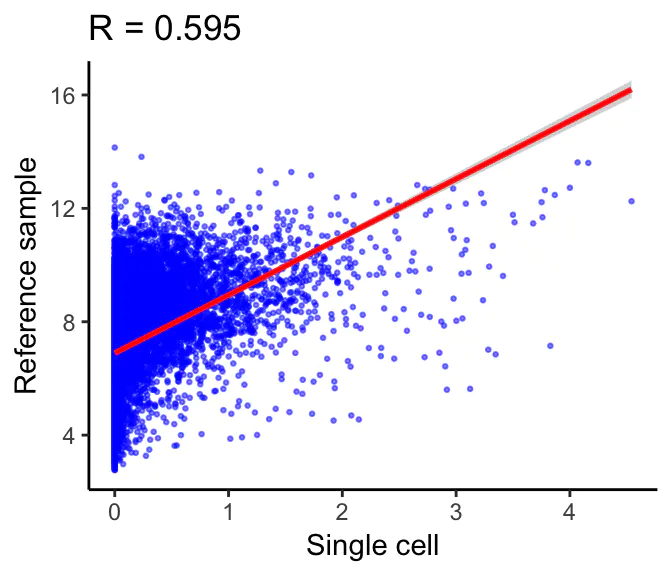
计算参考数据集中每个样本的单细胞表达的斯皮尔曼系数。相关分析仅对参考数据集中的变异基因(variable genes )进行。下面的示例显示了单个细胞(x轴)和参考样本(y轴)的表达式之间的相关性。这个散点图中的每个点都是一个基因
Step 2: Aggregation of scores by cell types
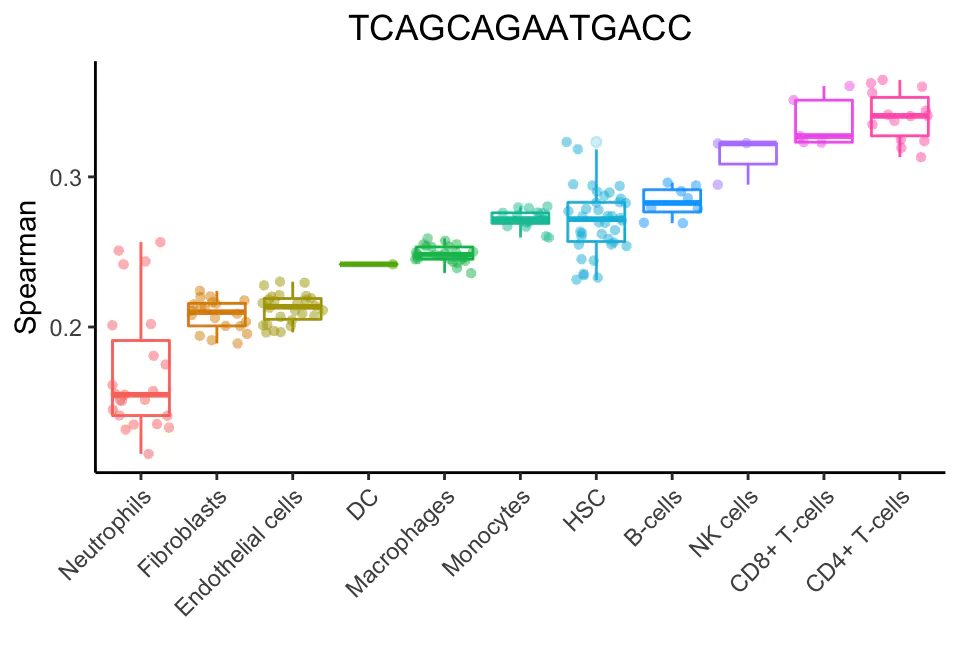
根据参考数据集的命名注释聚合每个细胞类型的多个相关系数,从而为每个细胞类型提供一个值。如上所述,这些示例是由广泛的细胞类型(“main”)或具有更高精度的细胞子集聚合的。默认值是每个细胞类型的相关值的80百分位数。
下面是一个针对单个人类细胞的注释过程示例。这里的点是使用一个细胞的所有参考样本(使用Blueprint+Encode参考)的Spearman系数。斯皮尔曼系数是按细胞类型聚合的(这里为了简单起见,减少了一组主要细胞类型)。每种细胞类型的单点评分是每个箱形图中的80%。这种细胞类型显然是t细胞或NK细胞,但不清楚到底是哪种类型。
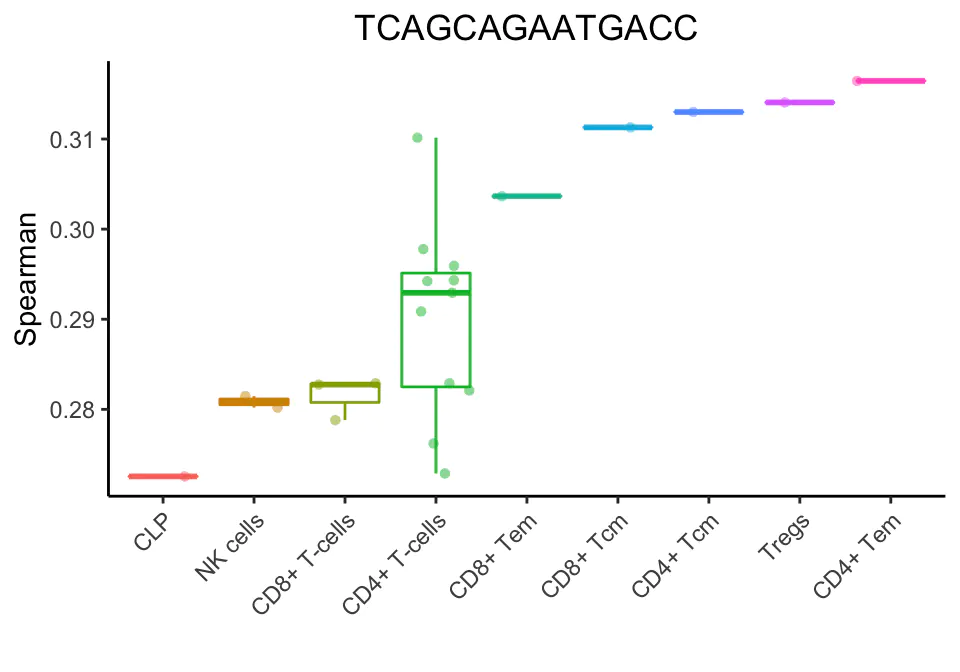
上面的分析将细胞子集和状态分组为主要细胞类型。SingleR允许更细粒度的细胞类型(只显示得分最高的细胞类型):
Step 3: Fine-tuning
在此步骤中,SingleR将重新运行相关分析,但只针对步骤2中的相关性较高的细胞类型。该分析仅对这些细胞类型之间的可变基因进行。移除最低值的细胞类型(或比最高值低0.05的边缘),然后重复此步骤,直到只保留两种细胞类型。最后一次运行后,与顶部值对应的细胞类型被分配给单个细胞。
在上面的例子中,SingleR清楚地表明了单细胞是一个记忆t细胞。然而,很难指出这些细胞子集中哪一个最适合它。微调步骤有助于分化密切相关的细胞类型。在第一次微调迭代中,选择顶部细胞类型(与CD4+ Tem评分相差0.05)。然后进行斯皮尔曼相关分析,但只使用这些细胞之间的可变基因。在对所有细胞类型进行微调之前,使用了3782个基因。在第一次微调迭代中,只有1819个基因被用来分化9种细胞类型。

在此迭代之后,将保留5种细胞类型。
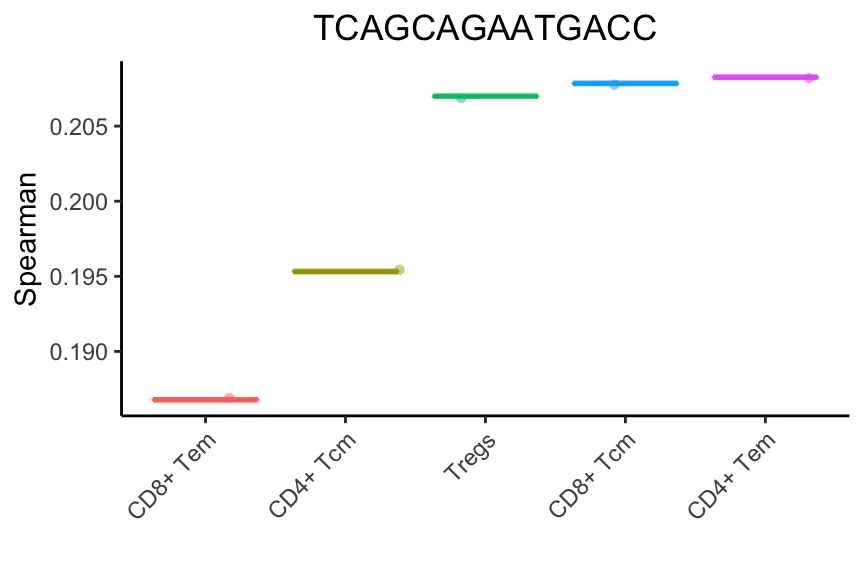
SingleR继续这些迭代,每次获的相关性最高类型或删除得分最低的类型。
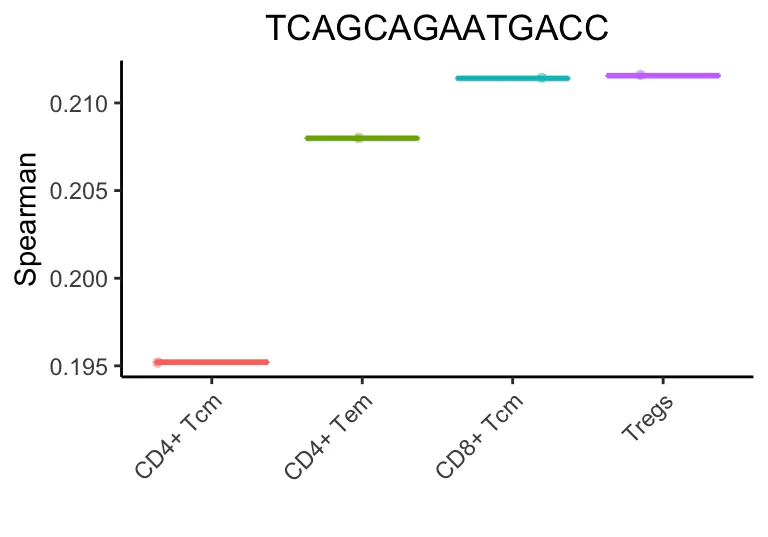

最后,成功的注释是一个调节性t细胞(Treg)。这个细胞实际上是一个排序的Treg,但是它不表达已知的标记(marker),如FOXP3和CTLA4,这使得基于标记(marker-based )的方法很难检测到。
二、安装
三、使用
四、具体案例
五、报错
六、讨论
参考资料
- https://www.jianshu.com/p/5dacf8afc9dc
- Nature Immunology volume 20, pages163–172(2019). Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. https://www.nature.com/articles/s41590-018-0276-y
- 使用文档:https://www.bioconductor.org/packages/release/bioc/vignettes/SingleR/inst/doc/SingleR.html#5_available_references
- https://github.com/dviraran/SingleR
