GATK MarkDuplicates 原理
1 为什么要进行 MarkDuplicates
重复 reads 被定义为源自单个 DNA 模板的序列。临床应用场景下比如检测肿瘤的突变频率,当测序的reads中 发生/不发生 变异是相互独立的事件时,计算出的突变频率才是准确的。但是PCR过程或者测序仪光学伪影导致的测序reads重复会打破相互独立这个条件,导致后续检测过程不准确。

2 导致 Duplicates 的原因
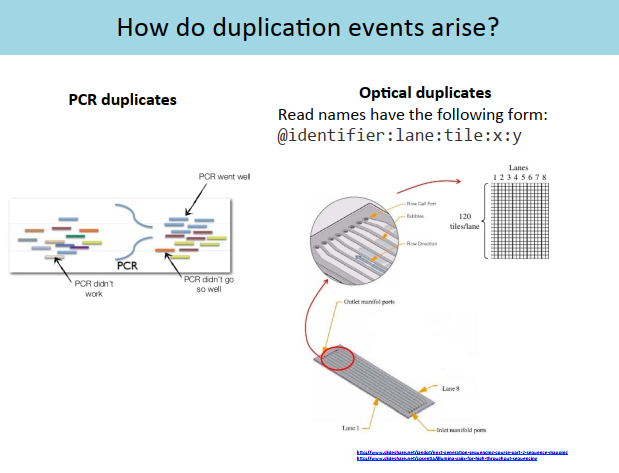
导致 Duplicates 的原因有两个:1、建库 PCR 步骤导致的 PCR duplicates;2、测序仪的光学传感器错误地将单个扩增的簇检测为多个簇,会导致 Optical duplicates。而且PCR 重复的比例会因测序量的增加而增加,光学重复是随机分布的。

3 怎样确认 Duplicates reads sets
MarkDuplicates 工具的主要原理是比较SAM/BAM 文件中 reads 的 5' 端的序列。Duplicates reads 集都是来自于同一个 DNA 模板,所以我们可以认为这些 reads 在基因组上有相同的5‘端起始位置。此外,BWA 在将 reads 比对到 reference 时,会在末端形成 “clips” ,这会使起始位置的情况发生变化。此外,当reads 比对到负链时,它的起始位置应该在3‘端。因此需要使用SAM 文件中的 flags(正负链) + CIGAR + start_pos 信息共同确认5‘端起始位置。
以下是不同情况下Duplicates sets判定:
(1)r1,r3,r5,r6是一个Duplicates sets,它们比对到了正链,起始位置都是1
(2)r2,r4是一个Duplicates sets,它们比对到了负链,起始位置都是7
(3)r7是一个Duplicates sets,起始位置是3

4 GATK MarkDuplicates 命令
MarkDuplicates 命令有很多参数,主要介绍一下几个参数:
--ASSUME_SORT_ORDER
可以接受 coordinate 排序输入,也可以接受 query 排序输入,但行为略有不同。当输入为 coordinate 排序输入时,比对上的 unmapped mates 不会标记为重复。但是,输入为query 排序输入时,比对上的 unmapped mates 会标记为重复。
--REMOVE_DUPLICATES=true
该参数为 true 时会直接删除duplicates序列,选择reads中碱基质量值加和最大的序列作为代表性序列。
该参数为 false 时会将重复标签加到flags中,其中1024代表重复序列(samtools flags 1024),GATK 后续处理软件会自动识别该参数,将其当做重复reads处理
-M 会输出一个 metrics 文件 用来记录单端和双端 reads 的重复数目
-I 输入BAM文件
-O 输出BAM文件
示例:
# markdup
/bioapp/java8/bin/java -Xmx16g -Djava.io.tmpdir=${outdir}/${sample}_tmp -jar /bioapp/gatk-package-4.0.11.0-local.jar \
MarkDuplicates \
--ASSUME_SORT_ORDER=coordinate --REMOVE_DUPLICATES=true \
-M=${outdir}/${sample}_gatk4.metrics \
-I=${outdir}/${sample}.sort.bam \
-O=${outdir}/${sample}_rmdup_realigned_recal.bam
五、 参考资料
- https://www.jianshu.com/p/21a64ea61792
- https://gatk.broadinstitute.org/hc/en-us/articles/360037052812-MarkDuplicates-Picard-
- https://hpc.nih.gov/training/gatk_tutorial/markdup.html
- https://cloud.tencent.com/developer/article/1626945
- https://qcb.ucla.edu/wp-content/uploads/sites/14/2016/03/GATKwr12-2-Marking_duplicates.pdf
这里是一个广告位,,感兴趣的都可以发邮件聊聊:tiehan@sina.cn
![]() 个人公众号,比较懒,很少更新,可以在上面提问题,如果回复不及时,可发邮件给我: tiehan@sina.cn
个人公众号,比较懒,很少更新,可以在上面提问题,如果回复不及时,可发邮件给我: tiehan@sina.cn
