【2.2.2】单克隆抗体的表征分析
在销售额方面,连续几年单克隆抗体药物在每年的销售额排名中一直是占据着前几名。有不下几十种新mAb药物产品和mAb生物仿制药正在开发中,并且每一个单抗药物都需要进行广泛详细的表征研究,以获得临床试验所需的批准并最终投放市场。
欧洲药品管理局(EMA)于2009年7月发布了有关mAb表征的监管文件。该EMA指南标题为“单克隆抗体及相关产品的开发,生产,表征和规格”。文件指出,研究者应详细的研究单抗的表征mAb。单抗表征应包括根据国际协调会议(ICH)指南Q6B(3)确定mAb的理化和结构性质,纯度,杂质等方面。
ICH Q6B为生物技术产品的表征提供了一套统一的国际公认原则,以支持单抗药物的市场应用。该文件建议进行分析以提供有关生物或生物制药产品的以下信息:
结构表征
- 氨基酸序列
- 氨基酸组成
- 末端氨基酸序列
- 肽图
- 巯基和二硫键
- 糖基化结构
理化分析
- 分子量大小
- 异构体
- 消光系数(或摩尔吸收率)
- 电泳图
- 液相色谱图
- 光谱
一、氨基酸序列和多肽测定
对于mAb,EMA指南和ICH Q6B要求通过适当的方法(例如,肽图分析,氨基酸测序,质谱分析)从DNA序列中推导氨基酸序列,并通过实验确认DNA衍生序列。另外,N-末端氨基酸序列(是否存在游离氨基酸或焦谷氨酸)和C-末端氨基酸序列(例如,存在或不存在C-末端赖氨酸)的变异性,重链等)应进行分析。
肽图分析(即使用mAb的特定蛋白酶消化酶,然后使用带有紫外UV和电喷雾质谱检测[LC/ES–MS的在线RP-HPLC分析产品)可提供有关分子量的信息。通过选择的蛋白酶从而使得mAb释放形成肽。获得的数据能够提供(或以其他方式)DNA衍生序列的确认,但不能提供mAb轻链和重链序列的确认。为了在一级氨基酸序列水平上提供确认,需要将许多蛋白酶消化物与在线RP-HPLC和串联MS/MS(LC/ES-MS/MS)分析联合使用。ICH Q6B实际上并不要求在一级氨基酸水平进行测序。但是,某些监管机构已采取了相应的行动规范。
由于质谱的测序无法(在大多数情况下)区分异亮氨酸和亮氨酸(这些氨基酸具有相同的分子量),因此要明确分配这两个氨基酸,就需要对纯化的肽在轻链和重链的可变区内进行自动N端测序。
二、氨基酸组成和消光系数测定
ICH Q6B指出:使用各种水解和分析方法确定氨基酸的总含量,并从所需产品或天然对应物的基因序列中推断出氨基酸组成(如有必要)进行比较。在许多情况下,氨基酸成分分析提供了一些肽和小分子蛋白质的结构信息,但是对于大蛋白质来说,这些数据的确定性通常较差。在许多情况下,氨基酸定量分析数据也可用于确定蛋白质含量。
如今,使用反相高效液相色谱(RP-HPLC)和柱前衍生化技术进行氨基酸组成分析,或者采用柱后衍生化处理的离子交换色谱法来确定生物制药产品的氨基酸组成。如果已知每摩尔蛋白质的氨基酸摩尔比和蛋白质的总分子量,则可以从产物水解物中检测到的每种氨基酸的量而计算蛋白质含量。同样,如果已知溶液的光密度(在280nm处)(用于分析氨基酸的等分试样)是已知的,则根据比尔朗伯定律(吸光度[OD] =ε.cl;其中ε是消光性)系数,c是浓度(M/L)。
三、末端氨基酸序列测定
进行末端氨基酸分析以鉴定mAb轻链和重链的氨基和羧基末端氨基酸序列。如果产品显示一个以上的末端氨基酸序列,则应确定末端的相对量。
使用十二烷基硫酸钠聚丙烯酰胺凝胶电泳(SDS-PAGE)分离轻链和重链后自动进行N末端测序,然后常规地印迹到聚偏二氟乙烯(PVDF)膜上,分析mAb轻链和重链的N-末端 。自动化的N末端测序使用Edman化学法,在切割N末端和随后的氨基酸之前,需要在蛋白质的N末端具有游离的氨基官能团以进行标记。这意味着对于许多mAb(在N端有焦谷氨酸,因此没有游离氨基),使用此方法的末端测序将不提供序列信息。在这种情况下,焦谷氨酰胺酶可用于在N末端测序之前酶促除去N末端焦谷氨酸残基。
没有类似埃德曼化学法(Edman化学)的完全可靠的方法来确定生物产物的C末端氨基酸序列。可以使用羧肽酶消化或质谱图策略获得与肽或蛋白质C-末端序列有关的信息。在后一种方法中,可以使用从肽图获得的数据和产物的完整分子量分析来评估蛋白C末端的完整性或参差不齐的末端存在。
四、巯基和二硫键测定
在肽图分析测序分析中,还应考虑游离巯基和二硫键。用特定蛋白酶消化后的肽图分析,然后在还原前/还原后使用在线LC/ES-MS或LC/ES-MS/MS分析,可提供全面评估二硫键和游离基所需的mAb中的数据。在碱性pH值下消化含有游离巯基的蛋白质时,必须小心,因为游离硫会在消化液中引发干扰,因此获得的结果可能与产品中真正的二硫键/游离巯基谱型不一致。
五、单抗糖基化结构测定
由于mAb是糖蛋白,因此还必须表征每种产品的聚糖部分。通常,mAb 在恒定片段(Fc)区域的每条重链中都有一个N链聚糖共有序列(天冬酰胺– Xxx –丝氨酸或苏氨酸,其中Xxx可以是脯氨酸以外的任何氨基酸)。mAb的轻链成分通常不会被糖基化。由于额外的N重链中可能存在连接的聚糖共有序列,应确认是否存在糖基化。ICH Q6B要求:对于糖蛋白,确定碳水化合物含量(中性糖,氨基糖和唾液酸)。此外,碳水化合物链的结构多肽链,寡糖模式(糖链分布)和糖基化位点尽可能分析。
如下图1所示,通常进行单糖成分分析以确定mAb的碳水化合物含量。液相色谱和气相色谱法(通常采用质谱检测)通常用于定义mAb碳水化合物的含量。所使用的方法应允许鉴定和定量中性糖(岩藻糖,甘露糖和半乳糖),氨基糖(N-乙酰氨基葡萄糖,N-乙酰半乳糖胺)和唾液酸(N-乙酰神经氨酸和N-羟乙酰神经氨酸)水平的产品在内。
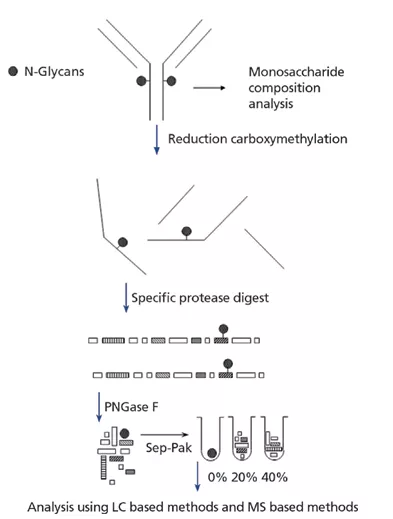
图1:用于分析单克隆抗体(mAb)糖基化的正常方法示意图。LC是液相色谱,MS是质谱。
在单糖成分分析之后,下一步分析将进入通常存在于每个重链上的寡糖的表征。上图1显示了用于从mAb 裂解,纯化和分析N-连接聚糖的方法学示例。
还原/烷基化和特定的蛋白酶消化旨在使mAb变性,以便相对较大的酶PNGase F可以有效去除N-连接的聚糖。一旦释放了N-聚糖,就可以使用简单的反相色谱柱将亲水性聚糖与疏水性更高的肽分离。的N端连接的聚糖馏分可使用质谱法或基于液相色谱进行技术分析。
通过分析mAb释放的N-聚糖(使用图1中所示的方法)获得的基质辅助激光解吸电离飞行时间(MALDI–TOF)质谱图如下图2所示。观察到的主要信号与常见的mAb聚糖G0F,G1F和G2F一致(见下图2)。
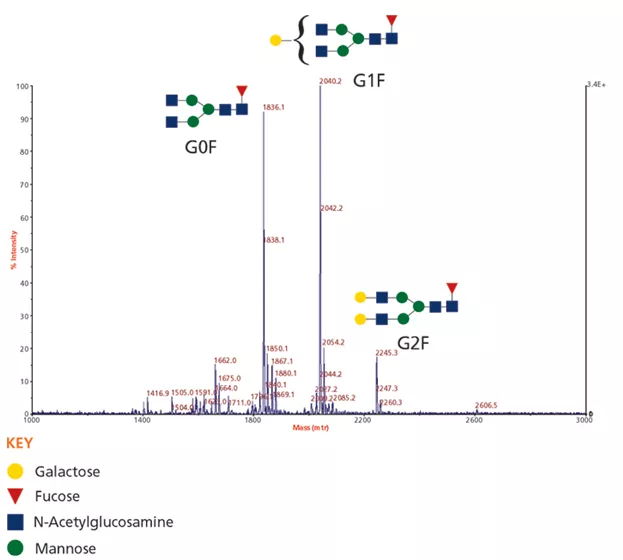
图2:从mAb释放的N-聚糖的基质辅助激光解吸电离飞行时间(Maldi-TOF)质谱分析获得的原始数据。在分析之前将N-聚糖过甲基化。
通过分析从同一mAb产品释放的N-聚糖获得的具有脉冲安培检测(HPAEC-PAD)痕迹的高pH阴离子交换色谱图如下图3所示。

图3:从高pH值阴离子交换色谱法获得的原始数据,并通过脉冲安培检测分析(HPAEC-PAD)测出mAb释放的N-聚糖。
两组数据都可用于提供所观察到的N-端连接寡糖的相对定量,并提示存在的N-连接聚糖的结构。糖基化是监管机构遇到的最常见的翻译后修饰。监管机构意识到,即使在制造过程中发生简单的变化,例如pH值变化或生长培养基的变化,糖基化也会受到显着影响。单糖成分分析数据(以甘露糖基化,半乳糖基化和唾液酸水平计)监管机构将通过对各种批次的mAb产品进行分析而获得的寡糖链分析数据用作评估制造商是否控制mAb制造过程的一部分。批次之间N-聚糖谱的可比性表明制造过程处于受控状态。
六、光谱测定
EMA和ICH指南还建议,应通过适当的理化方法来表征mAb的更高级结构。ICH Q6B建议使用“圆二色谱,核磁共振(NMR)或其他合适的技术”。如今,圆二色谱(CD)分析通常用于评估mAb的二级结构。从mAb的CD分析获得的数据如下图4所示。使用CDsstr软件评估原始数据可以估算各种二级结构的相对百分比,如下表I所示。
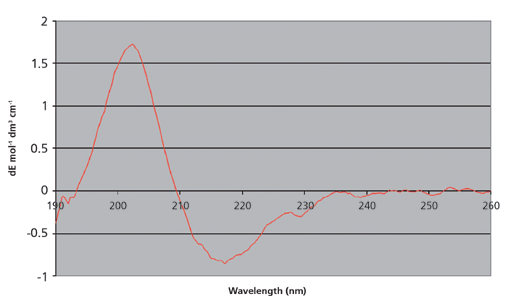
图4:从mAb的近紫外(UV)分析获得的原始数据。
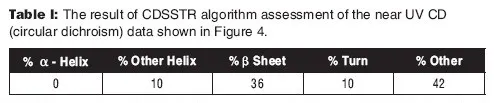
表I:图4所示的近UV CD(圆二色谱)数据的CDSSTR算法评估结果。
除了上面概述的结构特征要求外,还需要评估许多其他物理化学特性。
七、分子量大小
使用四极杆飞行时间(Q-TOF)仪器对完整和还原的mAb进行在线LC/ES-MS分析,彻底改变了mAb完整分子量以及释放的轻链和重链的测量方法。使用此技术从mAb的完整分子量分析获得的数据如下图5所示。
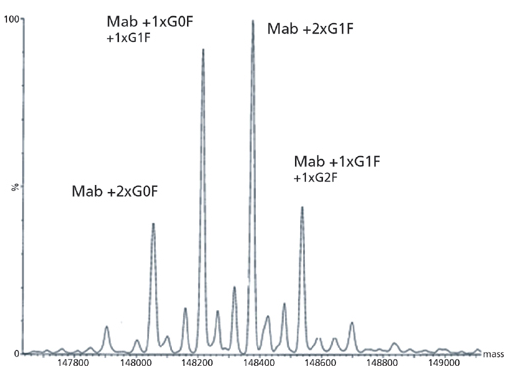
图5:通过在线电喷雾质谱检测(飞行时间定量)mAb的LC/ES-MS(Q-TOF)分析获得的去卷积质谱。

观察到的主要峰相距162质量单位,这与存在于分子的重链上的N-连接的寡糖的半乳糖基化的变化一致。还原后从mAb分析获得的质谱数据如下图6所示。
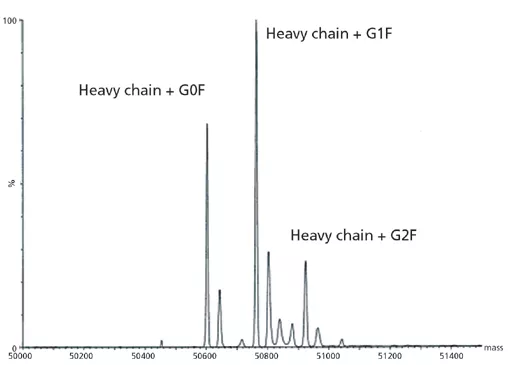
上图6A中所示的信号与轻链预期的信号一致,并且下图6B中观察到的信号与具有预期的N-连接寡糖的重链预期的信号一致。除了确定mAb的分子量以及轻链和重链成分外,这些数据还确认了蛋白质主链的完整性。
八、异构体
以类似的方式,成像毛细管等电聚焦(cIEF)带来了革命性的mAb异构体分析。cIEF在毛细管柱中是自由溶液的等电聚焦。诸如iCE280分析仪(Convergent Biosciences)之类的专用系统使用整个色谱柱紫外线吸收检测器来检测聚焦蛋白区域,从而避免干扰这些区域的聚焦。这项技术的独特之处在于它具有与传统凝胶IEF相当的分辨率,但结合了基于柱的分离技术的优势,包括定量(使用280nm的UV)和自动化。
从mAb的cIEF分析获得的原始数据如下图7所示。获得的数据可准确确定每种异构体的pI,并基于UV峰面积对每种异构体进行定量。

图7:单克隆抗体毛细管等电聚焦后获得的毛细管的UV(280nm)响应。
九、电泳和液相色谱图
通常使用等电聚焦(如上图所述),SDS-PAGE(还原前后)和毛细管电泳对mAb进行分析,根据大小和电荷提供产品分布。
使用尺寸排阻色谱法(SEC),RP-HPLC和离子交换液相色谱法进行色谱分析可根据尺寸,亲水性、疏水性和电荷分布提供产品表征分布。
电泳和色谱均提供有关产品身份,均质性和纯度的信息,并且经常用于纯化杂质,使用上述分析技术进行鉴定和结构表征。
十、蛋白质聚集
特别是对于mAb,必须使用多种方法评估每种产品中多聚体和聚体的存在。当前,监管机构通常要求使用多角度激光散射(SEC-MALS)和无柱技术(例如分析离心(AUC))对SEC进行交叉检查,以对从SEC分析获得的数据进行交叉检查。
由于聚体可能会因与SEC色谱柱的非特异性结合而丢失,或者实际上可能由于色谱柱上产物的稀释和剪切力作用而分散,因此需要使用无色谱柱技术确认SEC结果。
从mAb的SEC-MALS和AUC分析获得的原始数据分别显示在下图8和9中显示。在这种情况下,通过mAb的SEC-MALS分析获得的数据表明存在低水平的聚体(约2.6%)。从AUC分析获得的数据表明存在约5.0%的聚体。通常,使用AUC会比SEC-MALS观察到更高的聚集水平,这可能是由于可能降低上述分析数据中的聚集水平的因素所致。
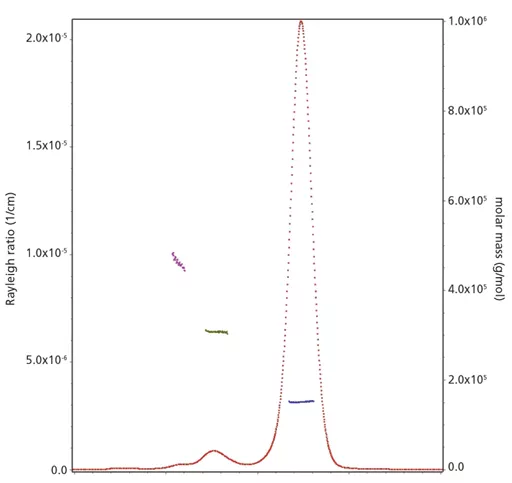
图8:通过大小排阻色谱对mAb进行多角度激光散射(SEC-MALS)分析获得的原始数据。

图9:通过mAb的分析离心(SV-AUC)分析获得的原始数据。
十一、结论
本文说明的技术用于整个产品有效期内的临床前阶段,以了解产品,以证明对制造过程的更改(例如按比例放大过程)不会改变产品的结构和理化性质,并提供数据以证明产品制造的一致性。同样,越来越多的特征分析技术(例如肽图分析和寡糖分析)被用作批放行测试方案的一部分,以将产品放行到临床中进行试验并最终投放市场。在开发任何mAb产品的过程中,至少要进行三批(通常是几批)的表征分析才能达到上述概述的程度。
总而言之,需要一系列分析技术来充分表征mAb产品。此外,要提交给监管机构,还需要对GLP和cGMP进行分析和验证。现在可以在几周内完成根据EMA和ICH指南对mAb进行全面鉴定,并且仪器的不断改进使分析实验室能够以更高的灵敏度,准确性和分辨率获得数据。分析的挑战始终站在这些技术的最前沿,并纳入越来越多的其他适当的技术,例如傅立叶变换红外(FT-IR)分析,以补充使用CD和二级结构分析,从而进行适当的汇总和总结。
参考资料
- 药时空 https://mp.weixin.qq.com/s/Pqz7Xb9vq-b6vylbmy6mjA
- IMS Midas and Knowledgelink; http://Pipelinereview.com/, Top 30 biologics 2010, special edition, Jan. 2011, http://www.pipelinereview.com/free-downloads/TOP_30_Biologics_2010_RDPN_Special_Mar_2011.pdf, accessed April 2011.
- EMA, Guideline on Development, Production, Characterization and Specifications for Monoclonal Antibodies and Related Products (London, Dec. 2008).
- ICH, Q6B Test Procedures and Acceptance Criteria for Biotechnological/Biological Products (1999).
