【4.5.7】长度无关的结构相似性丰富了抗体CDR规范分类模型(canonical class model)
摘要:
互补决定区(CDR)是组成抗原结合位点的抗体环。在这里,我们表明所有CDR类型都具有结构相似的不同长度的环。基于这些发现,我们为非H3 CDR创建了与长度无关的规范类。我们的长度可变的结构簇显示出很强的序列模式,表明它们是从相同的原始结构进化而来的,或者是某种形式的趋同的结果。我们发现,比长度无关的标准方法比长度相关的方法,不仅能将大量CDR聚类,而且还能更好地预测序列的规范分类。
为了证明我们的发现的实用性,我们从抗体库的3个下一代测序数据集中预测了CDR-L3序列的簇成员(超过1,000,000个序列)。使用长度无关的聚类,我们可以在结构上对另外的135,000个序列进行分类,这比标准方法提高了20%。这表明我们与长度无关的规范类别可能是抗体空间的高度普遍特征,并且可以大大提高我们准确预测通过下一代测序鉴定的新型CDR结构的能力。
缩写和首字母缩写词:
CDR,互补决定区;
PDB,蛋白质数据库
V区,可变区;
HMM,隐马尔可夫模型;
RMSD,均方根偏差;
DTW,动态时间规整; UPGMA,具有算术平均值的非加权对组方法(Unweighted Pair Group Method with Arithmetic Mean);
DBSCAN,带有噪声的应用的基于密度的空间聚类(Density-Based Spatial Clustering of Appli- cations with Noise);
OPTICS,确定聚类结构的订购点(Ordering Points to Identify the Clustering Structure);
AUC,曲线下面积;
ROC,Receiver Operating Characteristics
一、前言
标准抗体是Y型结构的蛋白质,由2条链(重链和轻链)组成。 它们是由免疫系统产生的,可以检测并作用于外来分子,也称为抗原。 抗体是研究最多的蛋白质类型之一。 自从1970年代解决了第一个抗体晶体结构以来,可用结构的数量呈指数增长。这种增长伴随着序列数据的相似趋势,导致建立了多个公开可用的序列数据库 ,目的在于收集和分析抗体测序实验的结果(例如,Kabat数据库, IMGT/LIGM-DB, abYsis,VBASE2、DIGIT7)。
抗体的结合特性主要由被称为互补决定区(CDR)的6个环的序列和结构决定。在轻链(L1-L3)上发现3个CDR,在重链(H1-H3)上发现3个。由于CDR的重要性,已经做出了大量努力来表征它们。抗体结构的比较表明,非H3 CDR(L1,L2,L3,H1,H2)仅形成相对少量的形状,称为规范类(canonical classes)。规范类描述了一组假设相似构象的环(loop),该构象由构成环的残基的数量和同一性以及与该环相邻的构架区中的一些残基确定。规范类别的理论假设,可以通过在特定位置存在一些“关键”残基来识别loop的类别。因此,使用规范分类(canonical classes,),应该有可能通过以下方式预测新的CDR的结构:使用其序列的关键特征对其进行分类。自从对Chothia和Lesk进行经典的分类研究以来,非H3 CDRs聚类为经典形式已经扩展了数次。
- 由Chothia和Lesk进行的CDR结构的最早聚类仅使用5种抗体结构进行,并且比较是手动完成的。
- 相比之下,Martin和Thornton创建了一种将CDR分类为规范形式的全自动方法,首先在扭转空间中对结构进行聚类,然后使用均方根偏差(RMSD)合并聚类。 Martin和Thornton也是第一个注意到规范模型的局限性的人,特别是序列并不是类群成员资格的完美决定因素。
- 在North等人的最新研究中,使用亲和力传播算法,将CDR结构聚集在扭转空间中[17]。此类群可作为联机数据库( http://dunbrack2.fccc.edu/PyIgClassify/ )获得。
还已经研究了仅涉及可用结构子集的规范形状。一些只分析了特定的链[12,20,21],而另一些只着眼于单个非H3 CDR,特别是CDR-L3。除了研究非H3 CDR的结构库外,还做出了巨大的努力了解CDR-H3的结构模式在Northwest等人的CDR聚类研究中,将CDR-H3的锚定区域(定义为环的前3个残基和后4个残基)分类为簇。
对CDRs(和一般抗体)的结构库的这些研究提高了我们从序列建模抗体结构的能力[32,33],为抗原识别提供了宝贵的见识[13,15],并启发了抗体设计的新方法。
在最早的聚类研究中,Chothia和Lesk注意到存在CDR loops,尽管长度不同,但彼此的结构比相同长度的其他CDR环在结构上更相似。 Martin和Thornton使用的聚类方法可以比较不同长度的环,但是作者发现的所有聚类仅包含一个长度的CDR。大多数后来的聚类是在不同长度的CDR在结构上是不同的假设下进行的。在这里,我们量化了不同长度的环之间的结构相似性,并创建了一种方法来查找与长度无关的CDR结构簇。我们表明,这些与长度无关的簇包含更多数量的独特序列,并且比与长度有关的对应簇更能根据序列预测结构。
后一个结果强调了这样一个事实,即不同长度的CDR之间的结构关系是基于序列模式的。使用与长度无关的结构簇,我们确定了不同长度的环结构之间相似的最常见原因。通过分析来自下一代测序数据集的CDR序列的簇成员关系,我们证明了我们研究的影响。我们表明,通过考虑不同长度环之间的结构相似性,我们能够将更多的CDR序列显着分类为结构簇(structural clusters)。
二、结果
CDR环的结构是从SAbDab数据库中可用的抗体结构中提取的,并按照“方法”部分所述进行过滤。使用动态时间规整(dynamic time warping,DTW)算法产生的结构比对,我们发现在所有CDR类型中,约50%的情况下,通过Chothia比对确定的插入位点在结构上都是正确的;在大约77%的情况下,正确的位点位于Chothia位点的一个残基内。
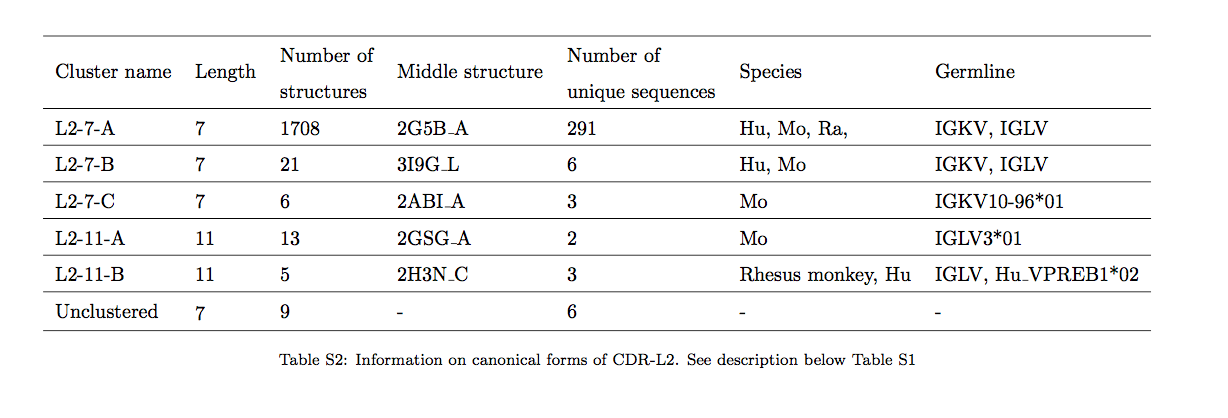
从我们的结构集中获取所有独特的CDR序列,我们使用DTW评分确定了与每个结构最接近的环(请参见材料和方法)。在所有CDR类型中,除了CDR-L2之外,对于CDR的某些部分,结构上最接近的伴侣(partner)的长度都不同(表1,图1)。该结果表明可以存在长度无关的规范类。


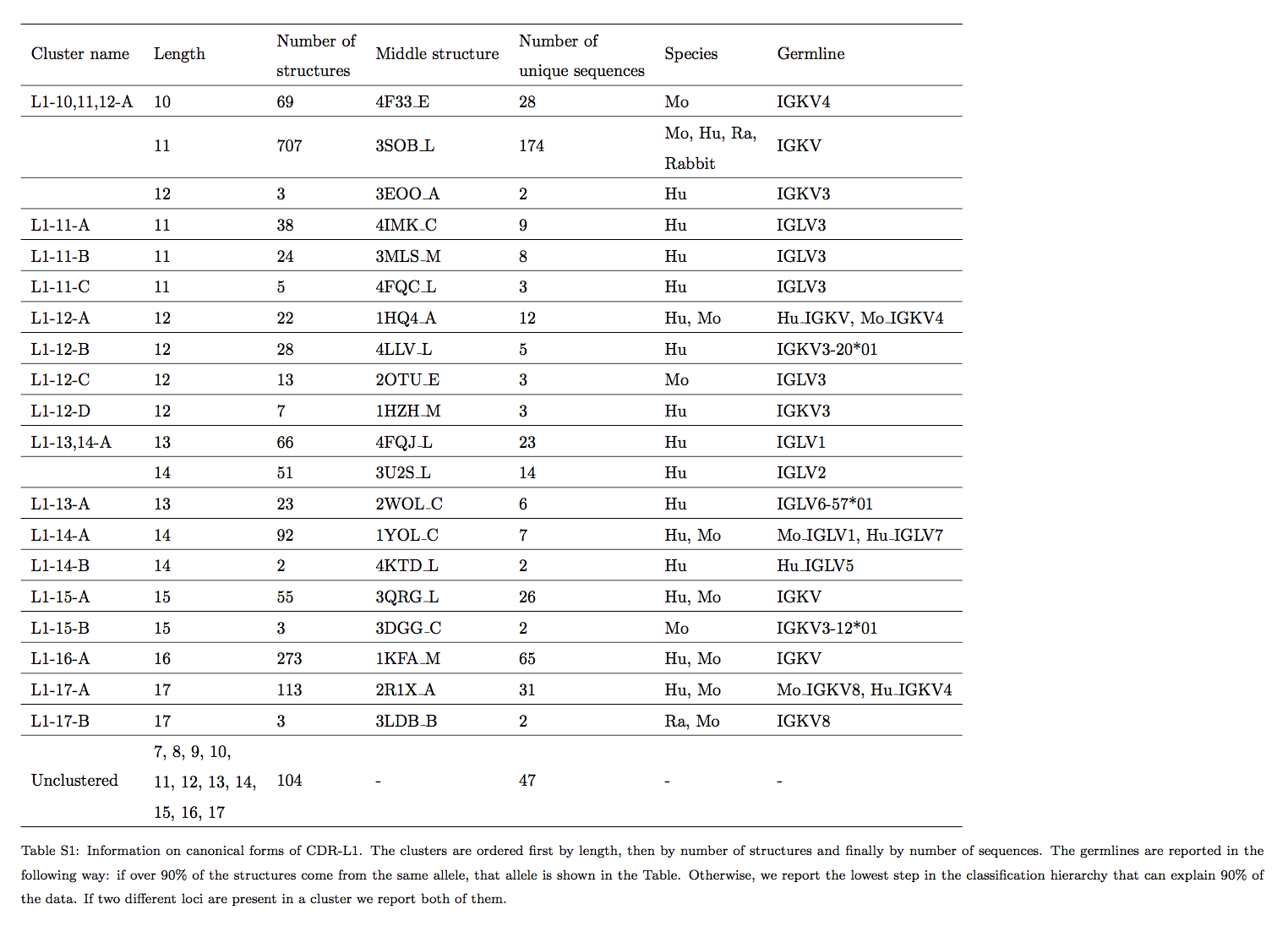
受此结果的启发,我们将基于密度的聚类方法和分层聚类方法(densitybased and hierarchical clustering methods )的思想相结合,以创建与长度无关的规范类。我们将所有CDR结构(不考虑序列冗余)用作聚类方法的输入(请参见材料和方法)。使用独立于长度的方法,我们总共发现了17个大簇,其中4个簇包含一个以上长度的CDR(要被分类为大簇,它必须至少包含6个独特序列)。表2总结了大型类群的结果。有关群集结果的详细说明,请参阅补充信息(SI)类群详细信息部分。
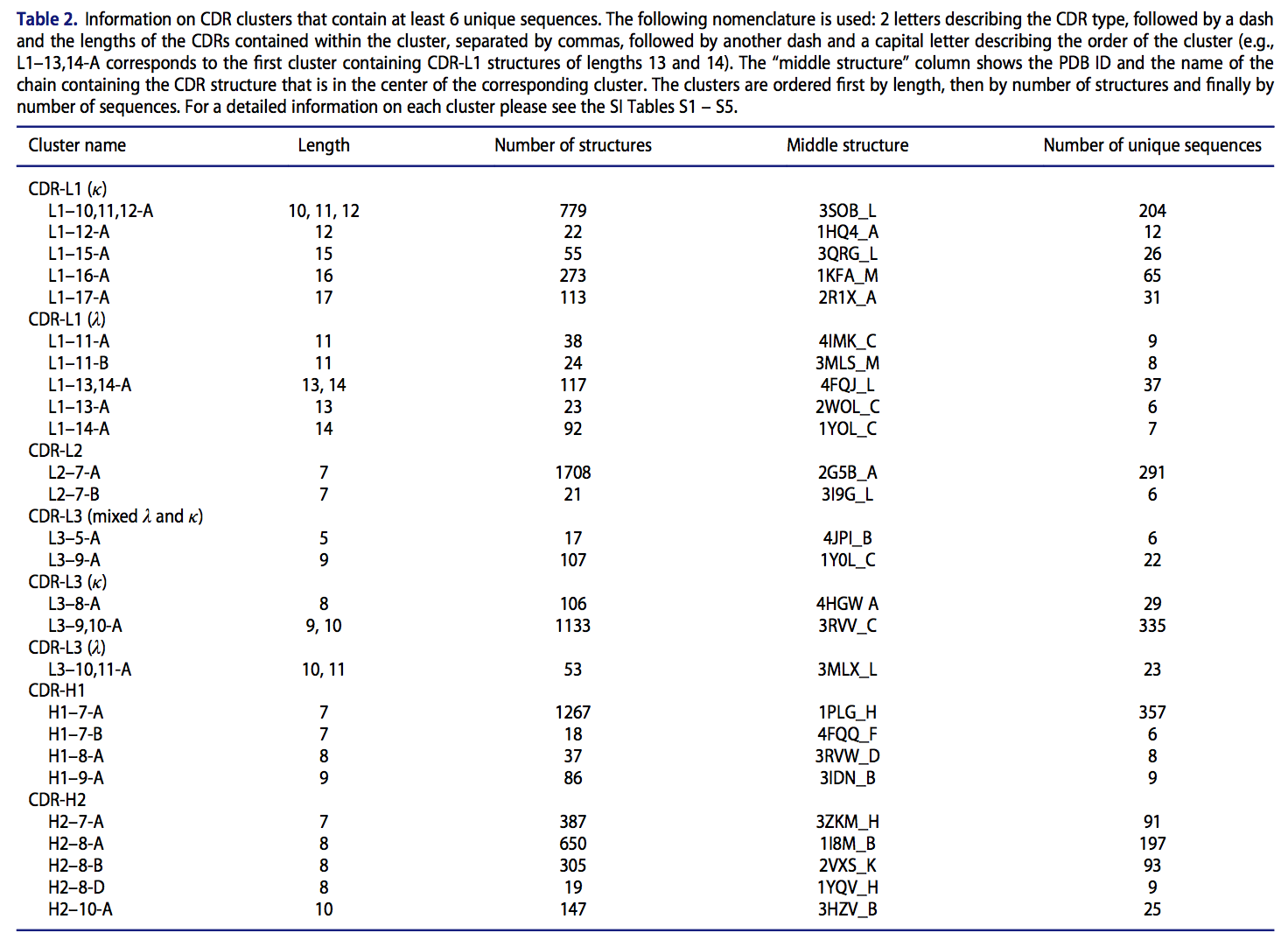
我们发现,大多数大型轻链簇仅包含k或λ轻链。 L3-5-A和L3-9-A是两个例外。 簇L3-9-A先前已经由North等人描述(作为簇L3-9-1)。 L3-5-A团簇包含的结构在North等人的工作中还不可用。 已公开,并且这些均来自广泛中和的抗体,这表明,不管链的类型如何,这样的环倾向于采取相似的形状。
我们对簇使用以下命名法:
- 2个字母描述CDR类型
- 后跟破折号和簇中包含的CDR的长度,以逗号分隔
- 后跟另一个破折号和大写字母描述簇的顺序( 例如,L1-13,14-A对应于包含长度为13和14的CDR-L1结构的第一簇。
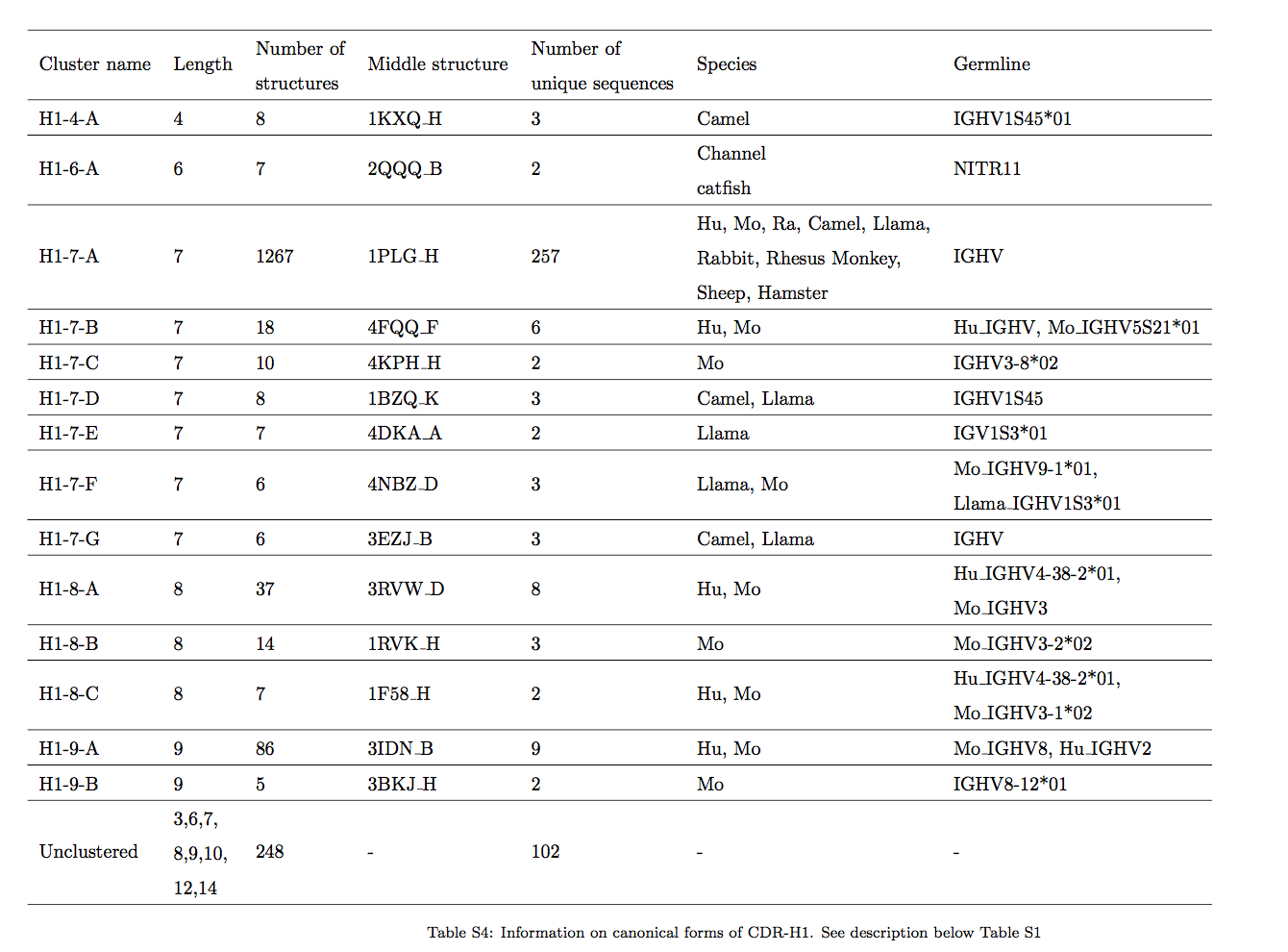

2.1 与长度无关的簇中的序列模式 Sequence patterns in length-independent clusters
为了使长度无关的结构相似性概念在loop模型中有用,不同长度CDR之间的结构关系必须通过序列相似性进行匹配。为了研究与长度无关的聚类是否包含清晰的序列模式,我们将预测方法的性能与聚类的与长度有关的版本进行了比较(请参见材料和方法)。我们发现,与长度无关的簇中增加的序列数提高了预测的精度。图2以CDR-L1簇 L1-13,14-A 为例说明了这一原理,该簇包含长度为13和14的λCDR。如果簇被长度分开,则预测精度会降低。长度为13和长度为14的CDR的序列徽标之间有明显的相似之处,特别是在Chothia位置29处存在Asn / Asp,这似乎是维持该簇中环结构的关键。
长度为10的CDR-L3(是簇L3-10,11-A的一部分)进一步说明了一致的序列模式的重要性。 这些CDR在长度为10的其他CDR-L3之间没有紧密的结构同源物,并且在聚类的与长度相关的版本中,没有聚类。 在群集的与长度无关的版本中,它们是群集L3-10、11-A的一部分,该群集主要包含长度为11的CDR。
为了评估在我们的集群上的预测方法的整体性能,我们绘制了每种CDR类型的接收器工作特性曲线(请参见SI图S6-SB)。 每种CDR类型的曲线下面积(AUC)均高于0.90(理想模型的AUC得分为1,而随机预测变量的得分为0.5)。
在下一部分中,我们将展示我们的聚类如何在CDR-L3 repertoire的下一代测序(NGS)的背景下改善预测。
2.2 下一代测序数据分析
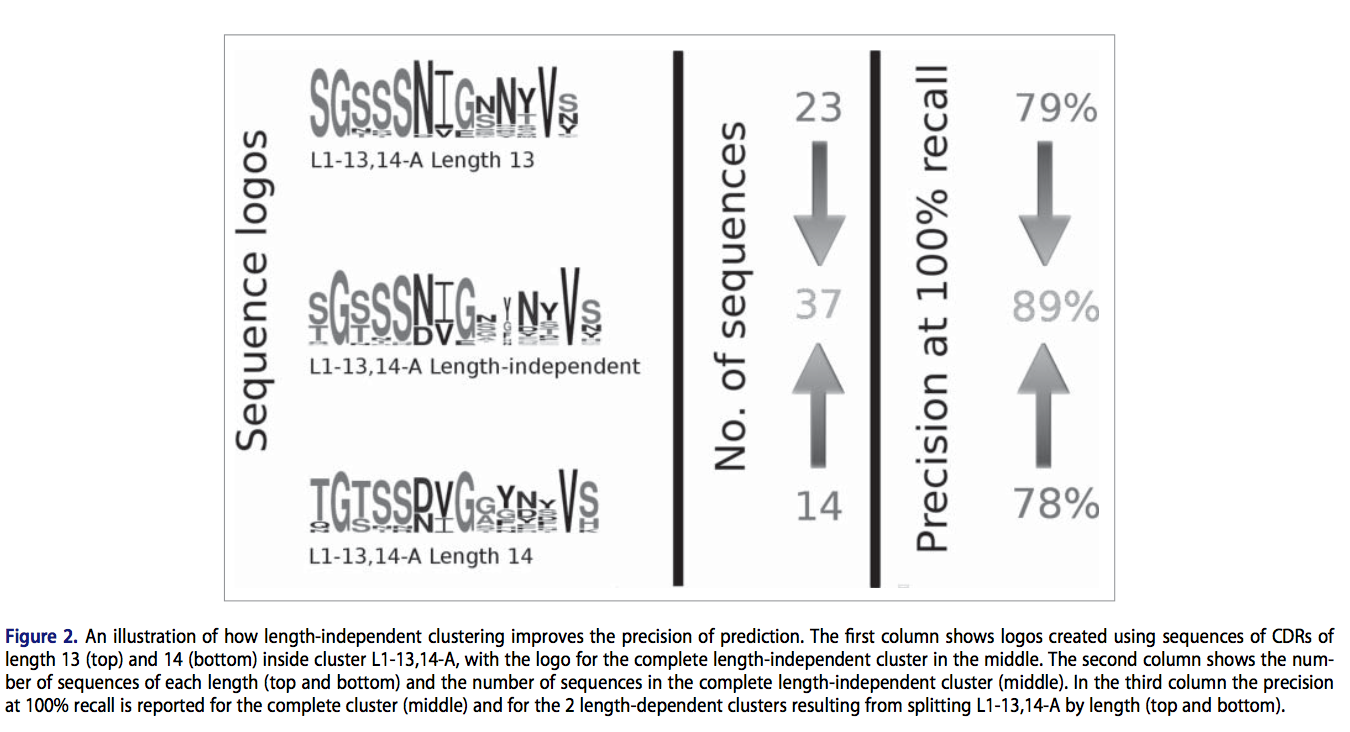
考虑到长度无关的簇包含这样清晰的序列模式,使其对预测有用,我们在考虑大型下一代测序(NGS)时,研究了结构集中显示的预测覆盖率的小增益是否具有重大影响组CDR-L3序列。我们检查了3个大型抗体NGS数据集:第一个数据集是通过UCB Pharma Ltd进行的测序实验创建的,包含超过~9,000,000条人类轻链序列;第二个数据集由DeKosky在2015获得,并且包含来自3个供体的198,148个人对配对的CDR-H3-CDR-L3序列;第三个数据集是从DIGIT数据库中提取的,由来自100多种不同物种的71,404条轻链序列组成。由于在所有数据集中只有CDR-L3序列可用,我们提取了这种类型的独特序列,从UCB数据集中获得了~1,000,000个序列,从DeKosky等人获得了 72,045个序列。数据集和DIGIT数据集中的12,960个数据集。
我们发现,这些数据集中的CDR-L3序列的长度分布与已知结构的CDR-L3的长度分布显着不同(参见SI图S3-S5)。例如,长度为10的序列占UCB数据集的26%(290,000个序列),而SAbDab数据库仅占6%。造成这种差异的主要原因是与NGS数据集相比,结构数据集中的k链相对丰富。结构数据集由大约78%的轻链和22%的λ轻链组成,而在NGS数据集中(仅包含人类序列),观察到47%的k链和53%的λ链分布更为平衡。但是,即使在按链型分离CDR-L3序列后,我们仍然观察到长度9的序列在结构数据集中被过度代表,长度10的序列被不足。由于这种差异,如果以长度相关的方式执行规范的类分配,则将更加困难。
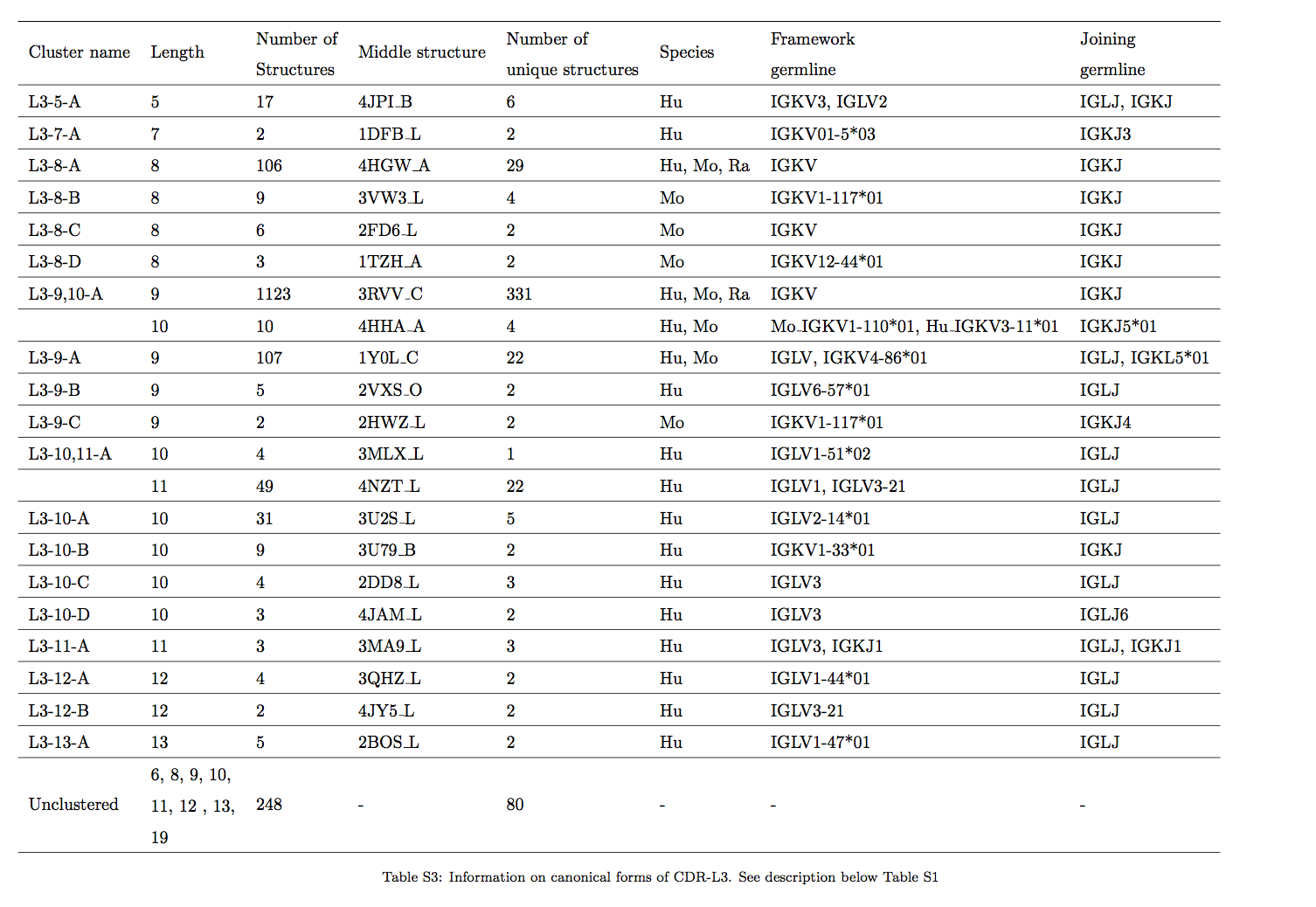
为了测试我们是否可以使用长度无关的方法为簇分配更多的序列,我们以预期的精确度在75%至90%(图3)之间,以长度依赖和长度无关的方式评估了独特CDR-L3序列的簇成员。 使用结构数据和HMMER返回的HMM分数估算了群集成员分配的精度(请参阅《材料和方法》)。 我们发现,在所有3个数据集中,我们可以使用与长度无关的方法来预测更多序列。 例如,以80%的精度,我们可以从UCB数据集中为集群分配额外的~125,000个序列(提高了21%,图3A),来自DeKosky等人数据集的8,958个序列(改进了21%,图3B)和来自DIGIT数据集的1,338个序列(改善了17%,图3C)。 总之,这些结果表明,使用独立于长度的聚类,我们可以在结构上表征抗体序列空间的很大一部分。
2.3 与长度无关的结构相似性的原因 Reasons for length-independent structure similarity
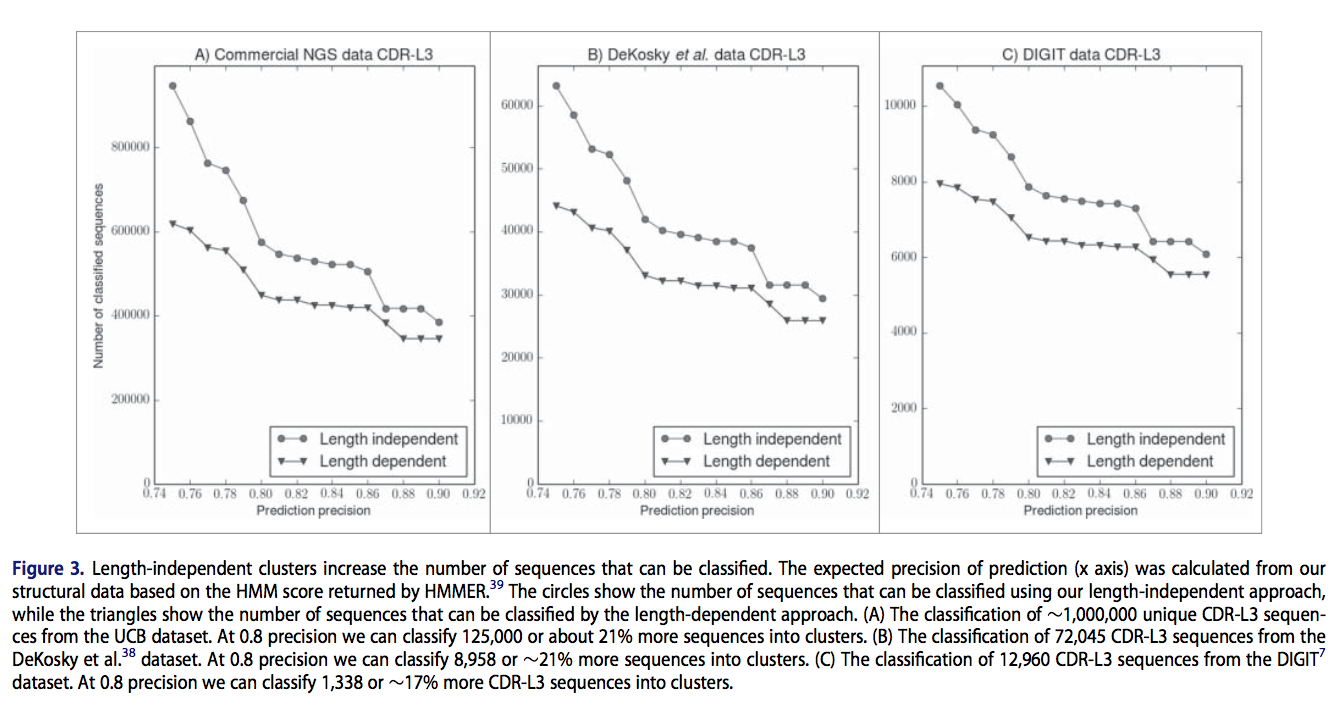
因为我们的长度不相关簇显示出很强的序列模式,所以我们研究了不同长度的CDR结构之间相似的可能原因。我们提出了三种自然机制,用于产生不同长度的结构和序列相似的CDR。
-
首先,种系包含大量的V区基因。不同长度结构之间相似的原因之一似乎是某些关键残基的同一性,这在不同种系之间是相同的(见图4A)。
-
其次,在研发的早期阶段,产生抗体的B细胞进行体细胞重组,在此过程中,V(可变),J(连接)以及(对于重链而言)D(多样性)基因片段被随机剪接在一起。这产生了抗体可变结构域的新序列。 VJ重组会影响CDR-L3的序列,这解释了为什么CDR-L3比其他轻链CDR类型更可变。我们发现,V和J基因的不同重排可能并不总是导致CDR结构发生重大变化,这可能导致不同长度的CDR-L3环之间的形状相似(图4B)。
-
第三,当B细胞被抗原刺激时增殖。在这种增殖过程中,重链和轻链的V区编码序列积累点突变的速率比其他基因高约一百万倍。少数表达抗体的亲和力更高的突变B细胞则进一步增加,刺激增殖。 此过程称为体细胞超突变(somatic hypermutation),可导致与靶标的亲和力增加1000倍。 在超突变阶段,可能会出现缺失和插入,尽管它们远不及取代常见。尽管体长不同,但由体细胞超突变产生的序列长度的变化可能会导致2个CDR具有相似的结构。在图4C中示出了这种可能的示例。
假设人类种系库包含~40种每种类型的功能可变基因(heavy,λ,k),每种类型的5个功能连接基因,23种功能多样性基因,并且N多样性和体细胞超突变可能增加了轻链和重链序列的数目约有1000倍,我们可以估计出人类有机体可产生约10种不同的抗体。我们在有限数目的抗体晶体结构中观察到长度无关的结构相似性这一事实表明,这在自然界中可能是相对普遍的现象。
2.4 重链互补决定区
尽管有迹象表明,天然抗体多样性的产生过程是观察到的长度独立的结构簇的主要原因,但我们在重链CDR中未发现任何长度可变的簇。在这里,我们更详细地描述了CDR-H1和CDR-H2的聚类结果,并讨论了明显缺乏长度依赖性结构相似性的背后可能的原因。
CDR-H1环的长度为3至13个残基。大多数结构(87%)的长度为7。共有14个簇,但实际上所有此类人类和小鼠CDR都集中在4个最大簇中(H1-7-A,H1-7- B,H1-8-A,H1-9- A)。观察到的长度和结构变异性似乎主要来自仅由重链组成的骆驼抗体的结构。
CDR-H2的长度多样性相对较低-在我们的结构数据集中仅观察到7至12个残基之间的长度环。大多数CDR结构(包括驼峰结构)都包含在5个最大的簇中(H2-7-A,H2-8-A,H2-8-B,H2-8-D和H2-10-A)。集群H2-7-A和H2-8-A中的环路结构相似,但不足以属于同一集群。
先前对CDR结构的分析讨论了Chothia 71位的构架残基如何影响CDR-H2的构象。 我们分析了整个大型簇中残基71的氨基酸分布(如图S2中的SI所示),发现簇H2-8-B和H2-10-A中的框架序列在此位置对Arg具有明显的偏好, 我们还发现,与以前的工作相比,簇H2-8-A中的框架序列显示71位的Arg丰度增加(~5%等价的North等人群H2-10-1,~10%的H2-8-A),使得残基较难预测簇成员。
2.5 与先前集群的比较
如上所述,先前已经报道了CDR结构的许多长度依赖性簇。在本节中,我们描述了North等人将CDR和最近的CDR聚类为长度依赖的规范类之间的差异。包含完整比较的表在SI表S6-S10中给出。
从我们的工作到North等人,通常将大型簇(包含至少6个独特序列的簇)很好地映射,通常具有一一对应的关系。但是,由于方法或长度独立性方面的差异,某些群集被拆分或合并。例如,在North等人的工作中,来自我们的集群L1-10、11、12-A的长度为11的循环被分为2个集群L1-11-1和L1-11-2。North由于在第30位的单个残基的构象发生了变化,所以这不会导致环之间的RMSD变大,但是会导致二面角的变化很大,并且,正如North等人在二面角空间中的簇一样, L1-10、11、12-A中的11个CDR长度分为L1-11-1和L1-11-2。对于我们的集群L1-11-A和L1-11-B可以看到相反的效果。这两个群集的central L1 loops(分别来自4IMK_C和3MLS_M)相距1.5 A,但在轴向空间上足够接近以属于North等人群集L1-11-3。由于采用了与长度无关的方法,North等人[17]将某些类分解。例如我们的集群L1-13,14-A在North等人的研究中按长度分为L1-13-1和L1-14-2。
较小的集群(包含少于6个唯一序列)的映射不太好,在我们的工作中通常没有相应的集群来匹配North等人的集群。我们的工作与North等人的工作之间还有另一个区别。North等人使用了非冗余CDR集,滤出了多次分离的同一抗体的结构。我们观察到,这些相同的序列可以具有具有明显不同的环构象的结构(例如,具有序列TGTSSDVGGYNYVS的CDR-L1环在结构上已多次表征为结构1MCB,1MCC,1MCD,1MCE,1MCF,1MCH, 1MCI,1MCJ,1MCK,1MCL,1MCN,1MCQ,1MCR,1MCS,并且在不同的PDB ID之间的差异超过1.5A。因此,我们决定包括所有CDR结构,而不考虑序列冗余。通过这样做,我们避免选择由于晶体堆积或解决结构错误而没有代表性的结构。这种方法还使我们能够观察到可以以2个规范状态存在的CDR序列(请参见SI)。但是,这也会降低我们预测构象的能力,因为可以在2个不同的结构簇中找到相同的序列。
三、讨论区
我们分析了不同长度的CDR之间的结构相似性,并使用它们来生成与长度无关的结构簇。与常用的长度相关方法相比,我们生成的簇数量较少,包含更多唯一序列。这提高了我们通过单独的序列将CDR分类为簇的能力。
假设对于CDR的一部分,最相似的可用结构是不同长度的结构,并且这样的结构相似性通常与序列相似性匹配,开发利用此信息的CDR建模方法应该会大大提高预测准确性。
我们已经描述了天然抗体亲和力成熟(affinity maturation)过程如何产生具有不同长度但相似结构的CDR。由于这些过程产生插入和缺失的可能性相对较低,因此与长度无关的结构相似性也很少见。但是,我们相信,随着新抗体的晶体结构的出现,长度可变的簇将越来越普遍。
我们在CDR-L3序列的3个大型NGS数据集上测试了我们的方法,发现我们的长度独立方法比标准技术可将~135,000或~20%的序列分类为更多的簇。 我们还观察到CDR-L3长度在结构数据集和NGS数据集之间的分布存在显着差异。 这种差异以及结构数据集中λ和k链之间的不平衡,是增加人抗体序列空间结构覆盖率的主要障碍。
四、方法与材料
4.1 CDR定义的选择
在本研究中,我们对除CDR-H2以外的所有CDR类型使用了CDR loop的Chothia定义,其中CDR-H2也包括N末端之前的2个残基。 当我们测试Chothia定义的CDR在任一末端最多延伸3个残基会改变聚类结果,尤其是预测准确性时(请参见序列部分的聚类预测),便做出了这一选择。 长度的变化仅对CDR-H2的结果产生了统计学上显着的变化,从而提高了预测准确性。 Kabat-Chothia编号中每个CDR的结果边界如下:CDR-L1:24-34,CDR-L2:50-56,CDR-L3:89-97,CDR-H1:26-32,CDR-H2 :50–56,CDR-H3:95–102。
4.2 数据选择 Data selection
该数据集是根据截至2014年9月在SAbDab数据库中可用的1833个抗体PDB(www.rcsb.org)构建的( http://opig.stats.ox.ac.uk/webapps/abdb/web_front/Welcome.php )。从数据集中删除使用X射线晶体学以外的方法解决的抗体结构以及分辨率高于2.8 A的。 CDR环的结构是从其余PDB及其锚,N端前的5个残基和C端后的5个残基中提取的。如果CDR结构的环或锚定区域中缺少原子,或者包含B因子大于80或等于零的骨架原子,则将其从数据集中删除。多次去除相同抗体的结构而得到的具有相同序列的环没有被去除,因为它们可以具有不同的结构。
我们对结构使用以下命名法:抗体的PDB代码为4个字母,下划线和链标识符为例(例如7FAB_L对应于PDB代码为7FAB的抗体的链L)。
4.3 相似度计算
最初,所有类型的所有CDR(例如,L1)的锚都被叠加,而与长度无关(叠加的锚反映了环相对于抗体的其余部分如何定向)。为了计算CDR之间的结构相似性得分,我们使用了DTW算法。该算法使用动态编程来找到通过成本矩阵的低成本区域的最佳路径。当比较两个相同长度的环时,该算法返回循环主干原子之间的RMSD。当比较两个不同长度的环时,该算法计算与遍历成本矩阵匹配的残基的骨架原子之间的RMSD(该方法类似于序列比对的Needleman-Wunsch算法,除了得分是根据残基的骨原子之间的RMSD,而不是取自序列相似性矩阵)。
CDR结构的所有图像均使用PyMOL.52程序生成
4.4 集群管道 The clustering pipeline
为了确保发现的簇能够反映所有潜在的结构和序列模式
- 首先使用DTW得分作为结构之间的距离度量和具有1.5 A截断的算术均数非加权对方法(Unweighted Pair Group Method with Arithmetic Mean,UPGMA)算法对CDR进行聚类。
- 接下来,使用隐马尔可夫模型(HMM)评估了从序列预测规范形式的能力(请参见从序列部分进行聚类预测)。
- 最后,使用基于密度的带噪声应用空间聚类(Density-Based Spatial Clustering of Applications with Noise,DBSCAN)重新聚类了包含超过6个唯一序列但可以以低于75%的精度和25%的召回率进行预测的规范形式,选择使用排序点识别聚类结构(Ordering Points to Identify the Clustering Structure, OPTICS)算法的最佳参数(再次使用DTW分数作为距离度量)。
- 之所以选择6个序列,是因为在较小的聚类中预测结果不可靠。结果参数显示在表3中。使用DBSCAN和OPTICS进行此重新聚类是为了确保每个聚类在结构上都是连贯的,如果数据允许,序列也连贯。

为了确保准确性不降低,我们将使用与长度无关的版本与长度无关的群集进行交叉验证,该版本使用相同的方法,参数和验证方法创建。使用HMMER预测器,可以针对每个聚类在一系列不同的HMM得分阈值范围内计算出正确率(TPR)和错误率(FPR)。 TPR和FPR在我们的集群中进行了宏观平均,并分别绘制了每种非H3 CDR类型的接收器工作特性(ROC)曲线,对于CDR-L1和CDR-L3,则分别绘制了长度独立型和与长度有关的版本。为了测量与长度无关的和与长度有关的ROC曲线之间差异的统计显着性,从TPR和FPR数据中采样了1,000个自举重复样本,并为每个ROC重复样本计算了曲线下面积。所得的平均值和标准偏差用于计算AUC差异的p值。发现曲线之间可能没有差异(CDR-L1曲线和CDR-L3曲线的p值分别为0.48和0.07)。所有群集的ROC曲线以及CDR-L1和CDR-L3的长度依赖性版本和长度不依赖性版本之间的比较在SI图S6-S8中显示。
4.5 序列聚类预测 Cluster prediction from sequence
为了从序列中预测规范形式,遵循了留一法交叉验证程序。首先,从每个簇中去除相同的CDR序列。然后,随机选择一个序列,并将其从每个簇中删除。使用程序HMMER 3.0从剩余数据为每个聚类构建隐马尔可夫模型(HMM)。最后,从聚类之外的所有序列为每个聚类构建背景分布HMM(以使用自定义背景分布,而不是一个用HMMER硬编码的代码,对HMMER源代码进行了修改,以返回“原始”对数似然率,而不是返回已经减去背景分布的分数。针对包含相同长度序列的聚类对选定的序列评分,并将其分配给评分最高的聚类(一对多分类)。重复该过程,直到所有序列都已分类。遵循相似的步骤对包含少于6个唯一序列的簇中的循环序列进行评分,并对落在簇外的循环进行评分,但是在那些情况下,完整的序列数据用于为大型簇创建HMM。
为了可视化用作HMM输入的CDR簇的序列模式,我们使用Weblogo软件包( http://weblogo.berkeley.edu/ )生成了序列徽标。补充信息中显示了包含至少6个唯一序列的簇的序列徽标。
4.6 遗传数据 Genetic data
当没有相应的IMGT条目时,从IMGT数据库( International ImMunoGeneTics information system, http://www.imgt.or )和SAbDab数据库中提取物种和种系数据。 如果IMGT记录中的物种注释与PDB文件标题之间存在差异,或者如果报告了人类生殖细胞系的CDR属于主要包含小鼠抗体的簇(反之亦然),检查了与PDB条目相关的文章以了解CDR的来源。
参考资料
- Nowak, J., Baker, T., Georges, G., Kelm, S., Klostermann, S., Shi, J., … Deane, C. M. (2016). Length-independent structural similarities enrich the antibody CDR canonical class model. MAbs, 8(4), 751–760. https://doi.org/10.1080/19420862.2016.1158370
