【4.4.4】组合肽库(combinatorial peptide libraries)预测TCE
官网:http://tools.immuneepitope.org/mhcii/
背景: 先前已经表明,组合肽文库(combinatorial peptide libraries)是表征I类MHC分子的结合特异性的有用工具。与其他方法(例如库测序或测量单个肽的亲和力)相比,利用位置扫描组合文库提供了具有成本效益,定量且无偏见的MHC分子特异性的基线表征。
结果: 在这里,我们介绍了该技术在19种不同的人类和小鼠I类等位基因上的大规模应用。这些包括特征非常明确的等位基因(例如HLA A * 0201),先前可用数据很少的等位基因(例如HLA A * 3201)以及先前关于特异性的报道相互矛盾的等位基因(例如HLA A * 3001)。对于所有等位基因,位置扫描组合文库能够阐明采用统一方法定义的独特结合模式,我们在此处提供了此方法。我们介绍了一种启发式方法,可将这些数据转换为主要和次要锚位置及其首选残基的经典定义。最后,我们验证了这些矩阵可分别用于识别牛痘病毒和流感病毒系统中的候选MHC结合肽和T细胞表位。
结论: 这些数据大规模证实了包括15个人类和4个I类小鼠等位基因的位置扫描组合文库方法对描述MHC I类结合特异性和鉴定高亲和力结合肽的功效。这些库显示出可用于识别特定的主要和次要锚位置,从而可简化基序,类似于其他方法所描述的基序。本研究还提供了可用于预测几个等位基因的高亲和力结合物的基质,这些等位基因以前没有结合特异性的详细定量描述,包括A * 3001,A * 3201,B * 0801,B * 1501和B * 1503。
一、背景
T细胞识别主要组织相容性复合物(MHC)分子和抗原肽或表位之间形成的复合物。 T细胞表位的鉴定对于促进免疫相关性的研究至关重要。不同的MHC分子与不同的肽结合特异性相关,通常称为MHC肽结合基序。大量文献涉及几种不同物种的I类分子的MHC结合基序的定义,包括人类,小鼠,黑猩猩和猕猴(例如,参见[1],以进行回顾)。通常,I类MHC分子识别长度为9至10个残基的肽,并在主要锚定位置携带具有相似理化特异性的残基。通常,主锚位于肽配体的位置2和C端,尽管已经描述了几个等位基因的其他锚排列。
可以使用多种不同的方法来定义MHC肽结合基序,每种方法都有其自身的优点和缺点。最常见的方法包括对天然存在的MHC配体进行池(pool)测序或评估单个肽库的结合能力。池测序方法是基于用MHC肽结合位点的酸性缓冲液洗脱后与MHC天然结合的肽的整体测序。这是一种非常简单有效的方法,已在数十个实例中成功应用[1]。它立即可靠地确定了MHC分子最主要的结合要求。该方法的另一个独特优势是基于生理处理的配体的表征这一事实。与该方法相关的缺点是它仅是半定量的,并且通常仅识别最规范的(严格的)基序。就利用这种方法进行表位预测而言,这可能是一个缺点,因为已显示许多显性表位不携带规范的池测序定义的基序。例如,原型显性人类白细胞抗原(HLA)A * 0201限制性流感病毒基质58-66表位(序列GILGFVFTL)[2,3]不包含与HLA-A * 0201池测序基序相关的主要锚模式,它指定了位置2上存在L或M。实际上,在最近的研究中,我们观察到已鉴定的HLA-A * 0201限制性牛痘衍生抗原决定簇中有57%(8/14)与A * 0201基序不符通过池测序分析得到[4]。
定义基序的最常见替代方法是基于体外建立定量MHC结合测定,然后测试一系列单个肽。这些肽要么是高亲和力结合表位或配体的单取代类似物,要么是无关肽的大型文库。该方法允许详细探测沿肽序列的每个位置的相对作用和化学特异性。当仅依靠单取代类似物时,对该方法的关注涉及以下事实:它可能反映了对特定的用作“野生型”的亲本肽具有特异性的结合模式,尽管在实践中,通过单取代分析鉴定的特异性模式通常非常吻合与其他方法论确定的方法[5-19]。相同的结合测定方法可用于测试给定大小的不相关肽(通常为100个或更多)的大文库,并且所有这些库都带有可接受的主锚残基。由于每个肽代表一个独特的序列,因此该方法克服了与单取代方法相关的担忧,即所识别的任何模式均取决于特定“野生型”配体的情况。
可以用不同的计算方法分析来自各个肽的亲和力数据,以得出定量基序,从而详细阐明结合能力的主要和次要影响(例如,参见[1,9,20-38])。基于此类数据的预测可以给出非常精确的肽结合定量近似值,并且可以区分带有相同主要锚定基序的候选配体。这种方法的最大缺点是,它取决于数百种等位基因特异性肽的检测方法。结果,这种方法可能是相对劳动密集的并且昂贵的。而且,肽序列的选择可以例如通过在特定序列位置上代表残基的上方或下方来将偏差引入训练数据中。
表征MHC分子结合特异性的另一种方法是基于位置扫描组合肽库的使用。这样的文库由大量不同肽的组合混合物组成,这些肽在特定位置均共享一个残基。测量此类文库的亲和力可有效评估共有残基对各种周围序列中结合的平均影响。因此,可通过测量一组180种混合物的亲和力来估算9个肽中所有20个残基的结合贡献。该方法已成功用于确定几种不同应用的特异性,包括与T细胞受体(TCR)识别相关的特异性分析[39],蛋白体切割[40]和与抗原加工(TAP)转运相关的转运蛋白[41]。 ],以及T细胞表位的鉴定[42,43]。在十多年前开始的几项研究中首先探讨了它们表征MHC结合特异性的功效[32,44-46]。已经发现,从组合文库分析得出的矩阵在预测具有高MHC结合亲和力的肽时表现良好[32,47,48]。 Buus在他对MHC研究的有远见的回顾中,提出了组合库的系统使用“人类MHC项目”,旨在完整地绘制人类免疫反应性[49,50]。
像单取代或肽库方法一样,从位置扫描组合库研究生成的数据提供了定量基序。使用位置扫描组合库的独特优势在于可以将其重新用于每个等位基因,从而可以节省大量成本。对每个等位基因重新测试相同的探针,也消除了将偏见引入被测配体组的风险。这些优势使我们能够系统地将组合文库应用于一组19个I类MHC等位基因。在这项大规模评估中,我们测试了这种方法是否在不同等位基因上均一地起作用。我们将其预测性能与生物信息学机器学习算法的预测性能进行比较。我们还开发了一种启发式方法,可将组合库亲和力数据转换为主要和次要锚位置的经典表示形式,从而使其与池测序中获得的结果直接可比。最后,我们在实际应用中测试了这些基质识别MHC结合肽和T细胞表位的能力。
二、方法
2.1 位置扫描组合文库和肽合成
如前所述[51]合成了组合文库。文库中的每个库均包含9个肽段,在单个位置带有一个固定残基。沿着9-mer骨架的每个位置上都代表20个天然残基,整个文库由180种肽混合物组成。
如其他地方所述[16]合成了用于筛选研究的肽,或者从Mimotopes(明尼阿波利斯,明尼苏达州/克莱顿,维多利亚,澳大利亚),Pepscan Systems BV(莱利斯塔德,荷兰)或A和A Labs(圣地亚哥, CA)。由A和A Labs合成了用作放射性标记配体的肽,并通过反相HPLC将其纯化至> 95%的同质性。使用分析型反相HPLC和氨基酸分析,测序和/或质谱测定这些肽的纯度。用氯胺T方法对肽进行放射性标记[52]。将冻干的肽以4–20 mg / ml的浓度重悬于100%DMSO中,然后在PBS + 0.05%(v / v)nonidet P40(Fluka Biochemika,Buchs,瑞士)中稀释至所需浓度。
2.2 MHC purification and peptide binding assays
。。。
2.3 生物信息学分析
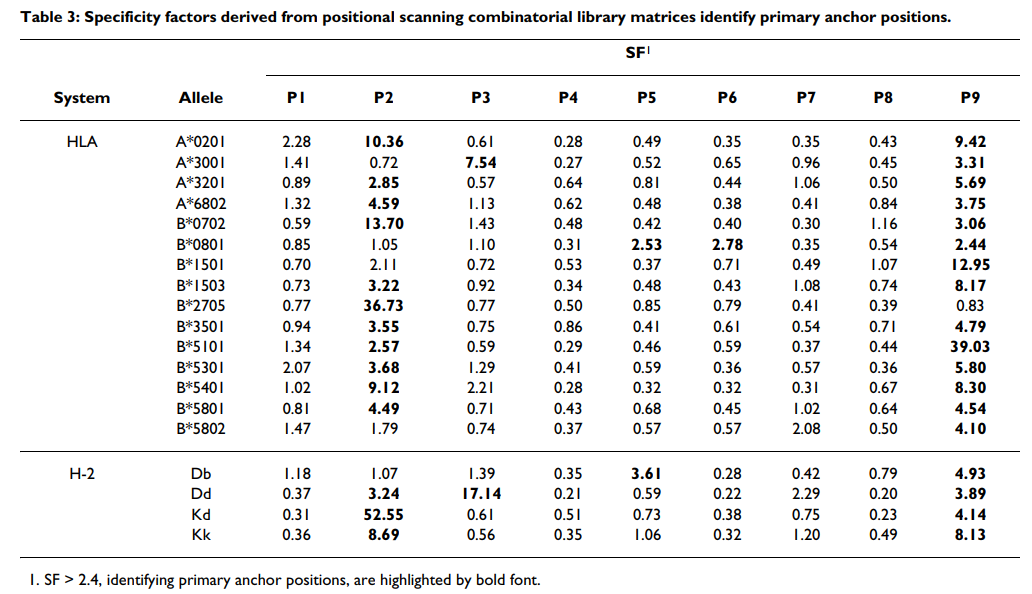
将每种混合物的IC50 nM值标准化为与整个180种混合物的几何平均IC50 nM值之比,然后如先前所述在各个位置进行标准化[17,18],每个位置对应于1,以使该值与最佳值相关。对于每个位置,计算平均(几何)相对结合亲和力(ARB),然后得出整个文库的ARB与每个位置的ARB之比。我们以这个比率命名,该比率将特异性位置上与所有20个残基相关的标准化几何平均结合亲和力不同于整个文库的平均亲和力的因素描述为该因子。根据计算,具有最高特异性的位置将具有最高SF值。然后将主锚位置定义为与SF> 2.4相关的位置。该标准确定了大多数残基与结合能力显着降低相关的位置。根据每个位置的残基比值的标准偏差确定二级锚。
为了鉴定预测的结合物,使用矩阵值对牛痘WR序列中所有可能的9-mer肽进行评分,其中每个肽的最终得分代表每个位置上相应残基的矩阵值的乘积。如前所述[56],使用稳定矩阵法(SMM)生成了通过组合位置扫描组合库和各个肽段数据集而得出的算法。
2.4 其他
。。。
三、结果
3.1 位置扫描组合文库方法预测HLA A * 0201结合肽的评估
先前在其他实验室中的研究表明,组合方法在预测几种鼠类MHC I类分子的结合物中表现良好[32,46,47]。为了验证对人MHC分子的作用是否相同,我们最初使用了位置扫描组合文库,其特征是特征最突出的人等位基因HLA A * 0201,其详细的主要和次要锚定基序已得到描述(例如,参见[3,9 ,16,34,61,62])。而且,针对该等位基因的几种不同的预测方法是广泛可用的,并且已经过严格的测试和比较(参见,例如,[36,47])。
以前,我们比较了A * 0201的几种预测方法的功效[47]。在该分析中,对免疫表位数据库(IEDB)[63-65]托管的3种算法进行了交叉验证,并通过对3000多个具有A *0201结合能力的肽进行了评分,直接对其他16种可公开获得的算法进行了评估。 。然后使用接收机操作员曲线(ROC)评估每种方法的性能,并计算曲线下的面积(AUC)。通过AUC测量的16种直接评估算法的性能介于0.935至0.788的最佳值之间(参见[47]和表Table1).1)。总体而言,平均表现为0.864,中位数为0.871。
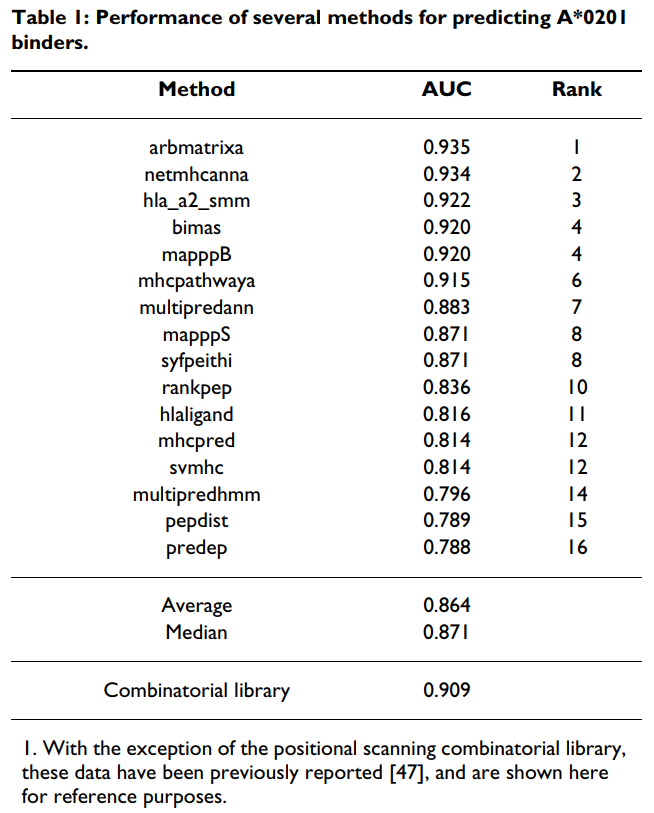
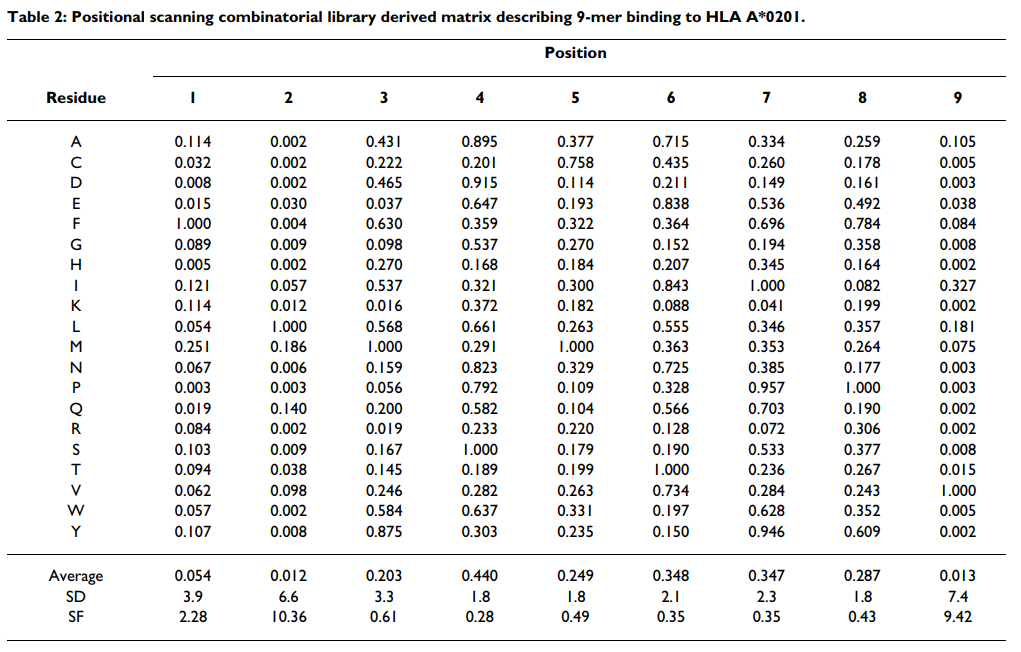
为了评估组合方法的相对性能,测试了每种9-mer混合物抑制高亲和力放射性标记配体与纯化A * 0201分子结合的能力。每种混合物测得的IC50 nM值在附加文件1 [请参见附加文件1]中显示。然后如材料和方法中所述将IC50 nM值归一化。所得的A * 0201基质(表(Table2)2)用于对与上面使用的相同的3089个9-mer肽进行评分。记录了预测的IC50和测得的IC50之间的良好一致性(r2 = 0.53),并且测得的AUC为0.909(表(表11和图图1).1)。该性能高于Internet上其他16种算法中10种的平均和中位数。该性能显着之处在于组合库仅使用180个数据点作为训练集。相比之下,使用训练集开发的性能最高的基于ARB,ANN和SMM的算法要多出10倍以上的数据点。
尽管对于其余算法而言,训练集大小的信息不可用,但鉴于A * 0201绑定数据的总体可用性,可以合理地假设这些算法已利用了类似大小的训练集。 此外,与比较中使用的大多数工具的情况不同,用于组合方法的训练集不会与测试集重叠,因此完全没有偏见[47]。 两者合计,本节中提供的数据进一步证明了使用位置扫描组合文库鉴定MHC I类结合肽的功效。
3.2 用于其他人和鼠I类等位基因的位置扫描组合文库矩阵的生成和验证
受到在A * 0201中获得的结果的鼓励,我们推导出了另外14种HLA(A * 3001,A * 3201,A * 6802,B * 0702, B * 0801,B * 1501,B * 1503,B * 2705,B * 3501,B * 5101,B * 5301,B * 5401,B * 5801和B * 5802)和4个鼠标(H-2 Dd,Kd ,Db和Kk)I类分子。 测得的IC50值在“附加文件1”中提供(请参见“附加文件1”),还将提交给免疫表位数据库(IEDB),以托管在IEDB分析资源中[66]。 如上所述将每种混合物的IC 50值归一化。 结果矩阵值在附加文件2中列出(请参见附加文件2)。 对于每个等位基因,矩阵确定了可重现的,特征性的结合模式。
3.3 通过位置扫描组合库方法识别主要和次要锚点位置
为了将组合矩阵的结果与合并测序和单个残基取代的结果进行比较,并有意义地总结每个计分矩阵中的大量数据,需要用简单的基序描述MHC结合。定义此类基序的第一步是鉴定对结合影响最大的肽位置。
和以前一样,A * 0201首先被用作模型系统。 A * 0201利用位置2和C端的肽残基作为主要锚点结合肽。在两个主锚位置,疏水或脂族残基是优选的或可耐受的。对结合能力的其他影响是由次要位置(最显着的位置1、3和7)上的残基引起的,在这些位置上可以注意到积极和有害的影响[9]。
为了得出可用于识别主要锚定位置的客观标准,我们认为与最高特异性相关的位置将与最低平均亲和力相关联,因为大多数残基都不被允许。因此,我们首先计算每个位置的平均相对结合亲和力(ARB),代表每个位置的值的几何平均值(见表Table2),2),标准化为整个文库的平均值。我们将此比率称为特异性因子(SF)。对A * 0201数据的分析表明,SF值≥2.4只能划定位置2和C端作为主要锚点位置(表(Table3).3)。为了测试SF方法用于识别主要锚点的一般适用性,我们为我们导出了组合矩阵的其他18个等位基因生成了SF值(表(Table3).3)。对于检查的18个其他等位基因中的13个(72%),SF> 2.4可以确定通过库测序或其他基序分析确定的所有主要锚点位置。总体而言,使用此标准可以确定先前确定的33/38(87%)个锚点位置。值得注意的例外是A * 3001和B * 0801。使用较高或较低的阈值会导致与先前描述的图案较低的对应关系。
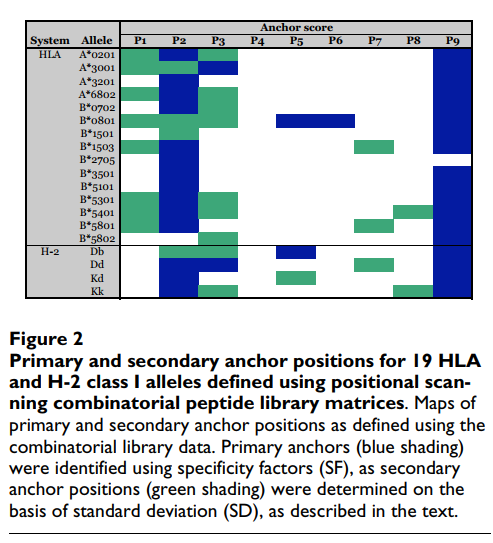
为了定义辅助锚位置,采用了不同的方法。 我们认为,与残基之间最高标准偏差(SD)相关的位置将对应于那些受到次级结合效应正向或负向影响最大的位置。 再次分析A * 0201数据,发现SD> 3可以成功识别先前描述为辅助锚的位置(图(图2).2)。 使用较高或较低的阈值会导致与先前描述的图案较低的对应关系。 将这一标准应用于其他等位基因,可以确定大多数等位基因的一个或多个主要二级锚位置(图(图2).2)。 在大多数情况下,发现主要的次要锚点位于1、3和7位。这种次要相互作用的模式与先前的分析基本一致[67]。
3.4 验证位置扫描组合库方法:识别主要锚点首选项
接下来,我们检查了在确定的主锚位置处的偏好模式与通过池测序或肽库方法鉴定的偏好模式相符的程度。再次从特征明确的A * 0201等位基因开始,我们发现相对亲和力在最佳残基的10倍以内的残基对应于先前描述的位置2和C端主锚上的优选残基(表22和and4).4)。更具体地,在位置2处,使用10倍阈值将L,M和Q鉴定为最优选的残基。相比之下,池测序方法确定了L和M。类似地,与C池测序中的V和L相比,组合文库在C端将V,I,L和A确定为首选残基。在这方面,组合库的10倍标准所确定的偏好大约在池测序分析所定义的更严格的基序与肽段筛选方法所鉴定的扩展基序之间的中途[16]。
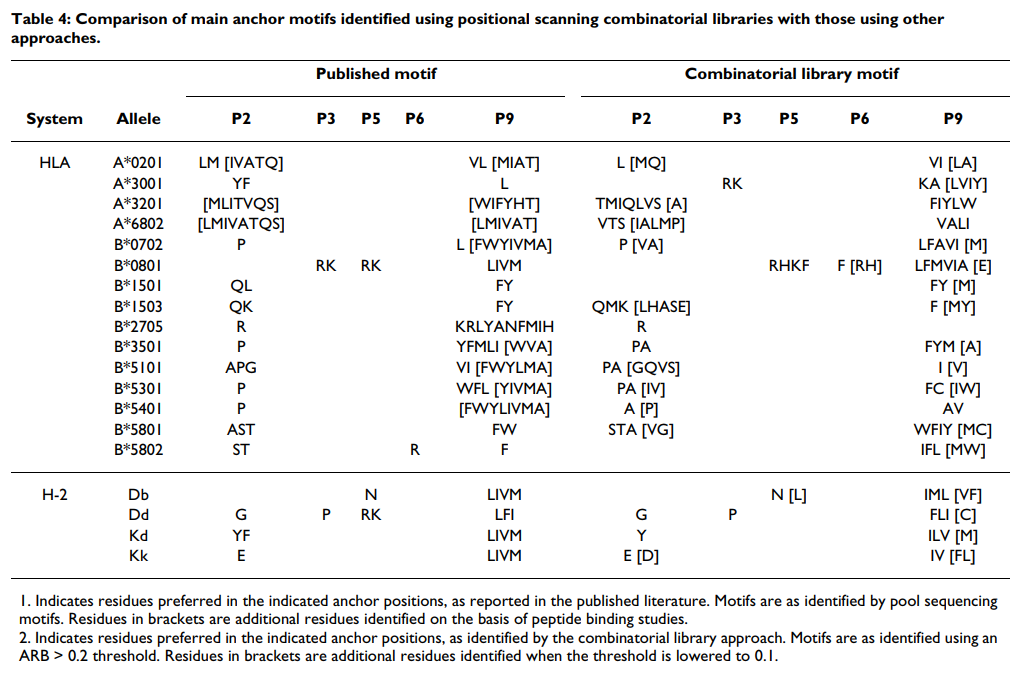
然后将相同的标准应用于18个其他等位基因。同样,组合库识别的模式在很大程度上遵循了先前描述的模式(表(Table4).4)。与A * 0201的情况一样,应用于组合文库数据的10倍标准往往比通过池测序确定的基序更广泛。然而,当使用更严格的阈值(例如5倍)时,鉴定出与池测序所描述的非常相似的更窄的基序。
该分析显示了几个意外的名称。将位置3识别为A * 3001结合的主要锚点,而不是位置2,与已发表的文献不一致,但是基于使用单个氨基酸取代肽的分析,这并非完全出乎意料(Sidney和Sette,未发表的观察结果) 。正如在库测序中所表明的那样,位置2中的优先级在这里被确定为主要的二级锚,似乎更倾向于小残基(V,T和A),而不是芳香族残基,尽管这些残基仍然可以很好地耐受。发现在位置3的偏爱碱性残基。池测序表明对C端疏水残基的偏爱。虽然组合文库产生的基序与这种一般特异性并不矛盾,但像对K的偏好那样识别A3超型是出乎意料的。然而,其他人(Harndahl和Buus等人,IEDB提交1000945,[63])和我们(Sidney和Sette,未发表的观察)随后的MHC肽结合研究证实了这种偏好。发现位置1和2是主要的辅助锚点。考虑到在池测序和位置扫描组合文库之间鉴定出的差异,该观察结果表明A * 3001可能能够使用多种不同的锚定排列结合肽。
在其他情况下,特别是B * 1501,B * 5802和B * 2705,在N-或C-末端均未定义明确的锚,这是更明显的化学特异性。 当使用池测序方法时,对于多个等位基因,也发现了在一个以上的锚残基处不能识别显性信号的类似失败(例如,参见[68])。
据我们所知,A * 3201的详细图案以前尚未提供。 已经提出,该等位基因将是A1-超型的成员,并且本文鉴定的基序与该关联一致。 然而,肽结合研究尚未能够证实该等位基因与该超型的其他等位基因具有显着的全部谱重叠。
3.5 位置扫描组合文库的应用:预测MHC结合肽
用5个选定的等位基因(A * 3001,A * 3201,B * 0801,B * 1501和B * 1503)进一步评估了组合文库矩阵的性能。这些等位基因中的每一个在人类中相对普遍,但是据我们所知,目前还没有大量高亲和力结合肽。使用这些矩阵,使用每个位置上相应残基的矩阵值乘积对牛痘Western Reserve(WR)株的所有9-mer序列进行评分。对于每个等位基因,合成大约对应于最高0.5%的前300个得分9聚体肽并测试结合。绑定数据汇总在附加文件3中[请参见附加文件3]。发现平均68%的所选肽以100 nM或更高的亲和力结合其相应的等位基因,在B * 1501的情况下最小为58%,在A的情况下最大为78% * 3001。相比之下,在A * 3001,B * 0801和B * 1501的情况下,可获取分数较差的肽组的结合数据,发现分数等于50%较低范围的肽仅粘合剂很少,结合率在1%至5%范围内(图(图33)。
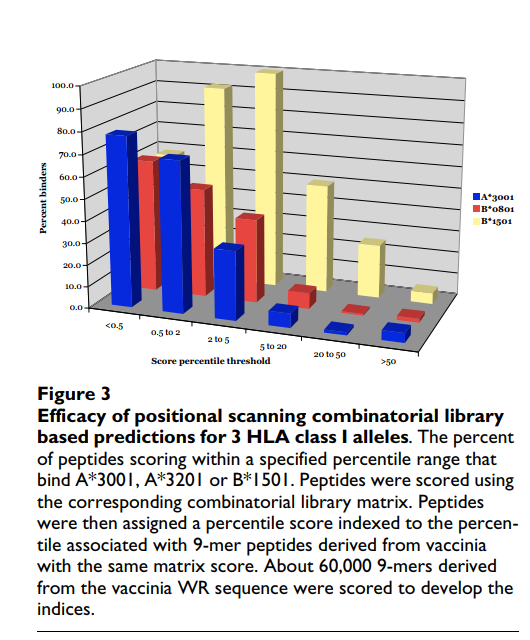
综上所述,这些数据进一步验证了组合库作为预测算法的基础的使用。 同样,本分析还提供了针对5个相对常见的I类HLA等位基因的,来自痘苗WR的高亲和力结合剂。
3.6 位置扫描组合文库的应用:预测T细胞表位候选物
我们最初想在经DryVax免疫的供体中测试从牛痘病毒中鉴定出的高亲和力肽组,类似于先前对携带来自常见超型的HLA等位基因的供体的调查[69]。 但是,在进行这项研究时,我们无法招募到足够数量的新接种的具有所需匹配HLA等位基因的供体。 相反,我们决定验证组合文库帮助从人类供体中识别的流感中鉴别T细胞表位的能力,为此我们可以将HLA等位基因A * 3001,A * 3201和B * 1501纳入多个供体。
为了充分利用组合库数据和这些等位基因的单个肽结合数据,我们利用了SMM(稳定矩阵法)方法[56],可以将这些数据组合起来以计算第二代矩阵。已经发现这些第二代矩阵比仅基于这两种方法的预测效果要好。使用第二代矩阵对A * 3001,A * 3201和B * 1501的第二代矩阵对代表性A型流感H1N1和H3N2菌株中存在的所有9-mer肽进行评分,如附加文件4中所示[请参见附加文件4],并为每个等位基因合成了得分最高的100个肽段。
测试了预测的高亲和力结合肽从具有匹配HLA的人类供体引发T细胞反应的能力。从白细胞或普通献血志愿者中分离出来自供体的PBMC,并通过高分辨率PCR进行HLA分型。该研究总共包括13位25-49岁的健康捐献者,其中3位A * 3001和A * 3201供者,8位B * 1501供体。用与供体单倍型相对应的组中的单个肽测定冻存的PBMC,并使用IFNγELISPOT测定法确定反应性。如方法中所述定义阳性表位。
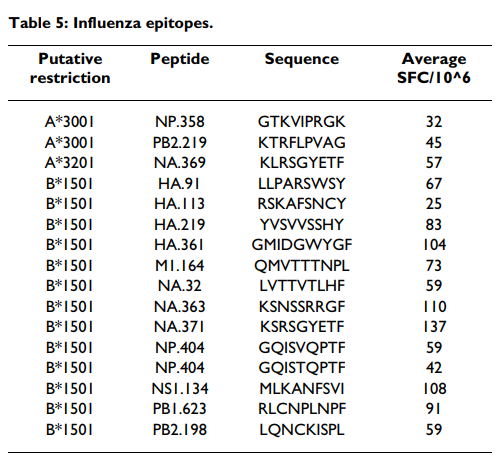
从这些实验中,成功地为每个等位基因鉴定了表位(表(Table5).5)。具体而言,分别在分别输入A * 3001,A * 3201和B * 1501的患者中鉴定出2个,1个和13个表位。但是,在一个以上的供体中没有发现任何肽。据我们所知,它们每个都代表新颖的表位,并且基于对A * 3001,A * 3201和B * 1501的预测,它们一起是第一批流感病毒衍生的表位。
四、讨论
由于肽与MHC的结合是引发T细胞反应的必要条件,因此预测肽结合的基于算法的方法通常被用作鉴定来自大型病原体的表位的第一步。在本研究中,我们利用9-mer位置扫描组合文库来表征几种小鼠和人类I类等位基因的肽结合特异性。当使用相应的位置扫描组合库数据生成矩阵来预测牛痘来源的结合剂时,发现在所有情况下,得分最高的0.5%肽中有58%至78%是高亲和力结合剂,具体取决于考虑了特定的等位基因。通过鉴定由人类捐赠者的PBMC识别的几种源自甲型流感病毒的表位,验证了源自组合文库分析的定量基序的生物学相关性。因此,这项研究提供了一组19种均匀生成的矩阵,这些矩阵可直接应用于预测MHC肽结合和T细胞表位候选物。
该方法的一个隐含特征是,它为所检查的每种MHC特异性提供了详细的定量基序。但是,通常用主要锚定基元的更简单术语概括MHC结合特异性通常是有用的。这种“极简主义”方法可以追溯到MHC结合的最早研究,在MHC结合中,使用库测序或单氨基酸取代分析确定了特异性。这些方法非常擅长表征等位基因特异性基序的最突出特征,并且所得基序通常形成了描述MHC结合的语法。为了扩展组合方法的实用性,我们开发了一种启发式方法,可以将组合库生成的矩阵数据转换为MHC研究的惯用语-更简单的主题。在大多数情况下,可以定义可概括的参数,以识别与其他方法所定义的主锚和副锚位置。
大多数HLA I类分子的结合特异性已通过晶体结构,池测序或肽结合研究进行了描述,该肽的主要锚定相互作用几乎总是涉及该肽的位置2和C端。对于迄今为止研究的大多数猕猴和黑猩猩I类等位基因,这种模式似乎也是正确的。正如A * 3001和B * 0801的情况所证明的,组合库分析表明,用于MHC肽结合的位置2 / C端锚间隔的范例并不总是正确的。以前在B * 0801 [70]的案例中已有报道,其中C端以外的位置3和5被确定为主要锚点。尽管通过组合分析没有重复这种精确的模式,但是本数据确实证实了肽中间带正电荷的残基对于赋予高亲和力结合能力的重要性。拾取MHC等位基因意外结合模式的能力是组合文库的关键优势之一,该组合文库没有事先预期哪些位置对MHC:肽相互作用可能很重要。
在我们之前的HLA超类型分类研究中[71],B * 0801被认为是离群值,因为它对于HLA而言是独一无二的,它使用位置3和5作为主要锚点位置。隆德[72]和赫兹[73]也做出了这个称呼。其他人[74,75]将其与我们[71]等人的等位基因分类,其他人[72,73]则指定为B7超型成员。在本研究中,组合文库分析表明位置5和6对于肽结合非常重要(除了C端)。因此,本分析没有提供足够的证据表明应将B * 0801分配给特定的超型,该超型主要由位置2(和C端)的特异性定义。我们可以注意到,脯氨酸,即B7超型相关的位置2特异性,确实在位置2中被B * 0801很好地耐受。同样,我们自己未公开的结合数据表明,B * 0702和B * 0801之间可能存在某些交叉反应。但是,目前尚未对这种潜在的交叉反应进行足够详细的研究以得出任何结论。
以前,B * 3501,B * 5101,B * 5301和B * 5401被分配给B7超型[71-73],B7超型[71-73]描述了一组共享脯氨酸优先位置2主锚的HLA等位基因。在本研究中,组合文库分析证实了这种偏好,但令人惊讶的是,还表明第2位等位基因对丙氨酸的耐受性良好。这表明至少在某些B7超型等位基因的库中可能存在重叠。 B7超型之外的等位基因,特别是与B58和B62超型相关的等位基因。尽管我们迄今为止所具有的结合数据表明,B7超型等位基因库谱重叠的大多数情况都将属于B7超型,并且脯氨酸是位置2的最主要偏好,一些交叉反应的证据也是很明显。实际上,最近的一项研究[76]发现与不同超型相关的等位基因之间的抗原决定簇具有高度的交叉识别能力。希望将来的研究将进一步阐明该问题。
在利用组合文库表征MHC特异性和鉴定结合物时,我们实现的方法在计算上很简单。我们已经为每个残基/位置坐标大量利用了相对结合值。为了预测结合剂,我们假设了肽侧链的独立结合,并将预测的结合倾向表示为每个坐标的乘积。还有其他方法可以处理原始数据,以生成预测矩阵或定义锚点位置。为了促进生物信息学界对预测方法的进一步研究,我们在此提供了十几种不同的HLA和4个H-2 I类等位基因的原始数据和处理后的数据。我们认为,该数据对于结合物和表位的预测将是有价值的,至少对于先前未详细表征的几个等位基因而言。
我们比较了组合文库与一组16种生物信息学方法的预测性能,这些方法用于最表征的人类MHC等位基因HLA A * 0201。尽管几种算法的性能优于组合库,但必须将其纳入考虑范围,因为这些算法基于多达十倍的训练数据。更令人惊讶的是,组合库仍然具有很高的竞争力,其预测质量优于16种算法中的10种。两者合计,组合库最小程度地提供了MHC结合特异性的非常可靠的基线表征,可以快速生成并具有成本效益。
使用基于组合文库的矩阵来识别候选肽集,可成功识别出A * 3001,A * 3201和B * 1501型患者的表位。然而,值得注意的是,在一个以上的供体中没有识别出任何肽。这种反应的多样性与先前在人类供体中将T细胞反应映射到牛痘衍生肽的过程相似。由于测试的A * 3001和A * 3201供体的数量很少,因此我们可能未估计阳性反应的数量。其他因素也可能是导致观察到的较低响应率的原因。所利用的供体库代表了近交群体,几乎所有的供体在A和B位点都是杂合的。因此,不同的供体反应可能反映了其他MHC等位基因在塑造总体T细胞库中的不同影响。类似地,未鉴定出多个供体中识别的显性表位可能是由于以下事实:供体集代表了暴露于不同病毒株的不同历史。
该研究的表位识别方面并未达到我们先前研究的水平和细节(例如[69])。造成这种情况的原因有很多,其中包括以下事实:所研究的等位基因并不罕见,但也不普遍,因此需要大量资源来确定足够数量的其他供体。结果,鉴定出的肽代表了初步性质的潜在先导。同时,数据确实证明了该研究中衍生的基质可用于表位鉴定,即使表位鉴定研究并不理想。此外,据我们所知,本研究中鉴定出的每个表位均代表新的表位,并且基于对A * 3001,A * 3201和B * 1501的预测,它们一起是第一组流感病毒衍生的表位。
五、结论
本研究从以前的研究[32,44,46,48,49]扩展了观察结果,显示了位置扫描组合文库对鉴定MHC I类结合肽的有用性。在此,我们已经为人和小鼠起源的19个I类等位基因提供了基于组合库的矩阵,其中包括一些以前未进行详细表征的等位基因。这些库也已显示出可用于识别特定的主要和次要锚位置,从而更简单的基序,类似于其他方法所描述的基序。对于A * 3001,A * 3201,B * 0801,B * 1501和B * 1503,已鉴定出以高亲和力结合的多组痘苗WR衍生肽。这些肽代表了未来研究的候选者,以鉴定来源于牛痘的抗原决定簇,牛痘是一种开发基于病毒载体的疫苗的重要病毒,此外还被广泛用作抗天花疫苗。最后,我们还确定了在HLA A * 3001,A * 3201和B * 1501供体中识别的几种流感病毒表位。
参考资料
