【4.3.1.4】疫苗表位预测工具
重组DNA的开发和测序技术带来了疫苗接种的新概念,其中已鉴定出能够刺激特定免疫反应的分离表位,并用于制备先进的疫苗制剂。替代由完整病原体配方组成的配方。在这种情况下,生物信息学方法在分析多个基因组以选择计算机保护性表位中起着至关重要的作用。可以设想,限定的表位或嵌合蛋白排列(包括靶表位)的混合物可以提供能够引起方便的体液或细胞免疫应答的原理设计。这篇综述介绍了最有利的在线免疫软件的全面汇编,并且可搜索,以便于疫苗的设计和开发。介绍了这些工具如何支持疫苗开发的前景。艾滋病毒和流感已被视为高变病毒疫苗接种方面有希望的发展的例子。还设想了该领域的观点。
一、前言
利用疫苗作为抵抗广泛传播疾病的工具,已经在对抗许多传染性疾病如流感,天花,水痘,百日咳,白喉,破伤风,脊髓灰质炎,肝炎和轮状病毒方面取得了长足进步[1],[2] 。包括减毒或灭活剂在内的常规疫苗可能需要长达15年的开发时间。这包括在适当条件下更大规模地培养所需微生物,以及有效灭活后评估疫苗的免疫原性。尽管这种疫苗挽救了无数生命,但它可能会产生不利的后果,如不良反应,诱发疾病或在某些情况下甚至导致死亡[3],[4],[5]。
生物信息学是一门科学领域,生物学,计算机和信息技术等多个学科融合在一起,以组织和存储由遗传学,分子生物学和生物技术产生的进步驱动的大量生物信息[6]。生物信息学的一个目标是有效,及时地简化和解释来自基因组,转录组和/或蛋白质组的信息[7]。该学科旨在促进健康利益,包括疫苗领域。
生物信息学工具的发展以及重组DNA技术(rDNA)的发展以及宿主免疫应答和病原体遗传背景的知识, 通过计算机计算机模拟预测来确定目标[8],将导致针对仅在一年或两年之内几乎没有或没有控制措施的疾病的新型疫苗,见图1。 通过rDNA技术开发的疫苗被设计为比传统疫苗更安全,更有效和/或更便宜。 为了实现这些目的,需要对病原体,特别是关键表位的充分了解,以诱导适当的免疫反应[9],[10],[11]。

尽管完整的基因组序列的可用性允许鉴定所有潜在的蛋白质产物,但该信息可能不足以鉴定实际上在病原体生命任何阶段表达的蛋白质子集。蛋白质组学在该领域也起着重要作用,因为它可以作为使用免疫组学技术鉴定和表征免疫原性蛋白质的基于基因组的方法的补充策略。疫苗组学,包括表征宿主对免疫的反应,提供了有关病原体与宿主细胞相互作用以验证候选抗原的有价值的信息。从这些学科获得的信息还加快了新抗原的鉴定和表征[12]。
在细菌物种上进行的蛋白质组学实验,不仅得到了从基因组测序和生物信息学分析获得的数据的验证,而且还导致了新蛋白质的发现,这可能是潜在的新疫苗候选物[12],[13],[14]。 Adamczyk-Poplawska发表了有关蛋白质组学对抗菌和抗病毒疫苗开发的影响的广泛综述[12]。
重要的是要注意,此处介绍的标准生物信息学网络工具不足以对整个基因组进行详细分析。免疫信息学是一门学科,其主要目标是使用计算和数学方法转换大规模免疫学数据,以理解和组织这些大规模数据以获得具有免疫学意义的解释[15],[16]。该领域的工具基于统计和机器学习系统,用于建模分子相互作用(例如抗原加工和呈递)的研究,并且在定义与理解免疫系统机制有关的新假设方面也发挥了作用[17], [18]。
这篇综述的目的是根据我们自己的经验以及对文献和网络数据库的艰苦搜索,为一些最有用的在线免疫学软件和可用于基因组分析的可搜索数据库提供一个门户,并就这些工具如何在疫苗开发领域,特别是基于表位疫苗的开发。
二、基于表位的疫苗
由于表位在疫苗设计,疾病预防,诊断和治疗方面具有巨大潜力,因此临床和基础生物医学研究人员都特别感兴趣。使用rDNA技术,我们可以分离出特定的表位,这些表位可以代替疫苗中的整个病原体。然而,在病原体的表位多样性中,重要的是要注意,并非所有表位,即使是那些似乎占优势的表位,在引发抗体产生的能力上也都相同[19],[20],[21] ]。
除了产生特定的免疫原而不是整个病原体之外,rDNA还允许进行合理的疫苗设计,包括产生嵌合蛋白,这为免疫原设计提供了多种可能性。包括具有以下优点的多表位疫苗的概念:单分子中包含几个免疫保护性表位,免疫优势但非保护性的表位被丢弃,而具有佐剂作用的表位(如混杂的T细胞表位)可以被包括在内以增强免疫原性[22] 。这些功能提供了设计多目标高效疫苗的可能性。然而,当遗传变异性与特定病原体相关时,设计此类免疫原的必要条件在于发现免疫保护性表位和变异体。
表位驱动的疫苗是一个有吸引力的概念,已在许多研究小组中成功应用,特别是针对针对可变或快速突变病原体中的保守表位的疫苗的开发[23],[24]。
理想情况下,疫苗中选定的表位应在病原体及其变体的不同阶段中保守。 此外,应考虑所需的免疫反应。 细胞毒性T细胞介导的反应是通过包含以线性表位为主要靶标的细胞内抗原加工的途径引起的[21]。 在这方面,选择用于疫苗的表位必须与一个以上主要组织相容性复合物(MHC)等位基因具有结合亲和力,并且必须覆盖主要人群[25],[26]。
包含许多由常见MHC等位基因识别的表位的蛋白质被称为混杂结合物(promiscuous binders)[26]。 人类白细胞抗原(HLA)超型是指一组具有重叠肽结合特异性的HLA等位基因。 给定的HLA超型中的等位基因通常代表相同的表位,指抗原表面上能够引发针对T细胞识别的免疫反应的区域[25],[27]。
另一方面,引发体液反应依赖于线性表位和构象表位的识别。 后者对嵌合疫苗设计构成了挑战,因为它们必须保留其天然构象才能发挥功能[28]。 因此,必须掌握有关整个抗原结构的知识,以帮助合理设计针对构象B细胞表位的疫苗[27]。
在这种情况下,生物信息学方法可有助于设计基于表位的疫苗。 使用这些工具,可以完成对表位的适当计算机选择[29],[30]。
三、生物信息学工具预测潜在的T细胞结合表位
将生物信息学应用于疫苗开发的第一步包括区分具有潜在免疫保护作用的表位与没有免疫保护作用的表位。 由于T细胞表位以线性形式与MHC结合,因此配体与T细胞之间的界面可以精确建模[31]。 当前众所周知,表位通过其R基团侧链和位于MHC底部的口袋之间的相互作用,共同连接到MHC I类和II类分子的结合槽中[32],[33],[34] 。 基于此知识,已建立了大量的T细胞表位映射算法,并用于开发工具以快速识别推定的T细胞表位[31],[35],[36]。
MHC-I结合预测因子目前非常有效,并且具有广泛的等位基因覆盖范围,已估计出90%至95%阳性预测值的预测准确性[29],[37],[38]。在众多用于MHC-1等位基因的服务器中,包括RANKPEP,它可以通过蛋白质序列或使用位置特异性评分矩阵(PSSM)进行序列比对,预测与MHC-1和MHC-II分子结合的肽。此外,它预测了其C末端可能是蛋白酶体切割的结果的MHC-1配体[39]。这是一个友好的平台,可为人类和小鼠的MHC-I和MHC-II等位基因(分别为118和67个等位基因)提供最广泛的等位基因覆盖。为了使用PSSM搜索表位序列中的MHC-1配体,使用了Python编写的动态算法。它会按照PSSM宽度的长度对所有蛋白质片段进行评分,并对其进行相应的排序。评分从每个序列的开头开始,PSSM每次在序列上滑动一个残基,直到到达序列的末尾。此外,为了从分级肽的列表中缩小潜在的结合物,将结合阈值定义为包括PSSM中90%肽的得分值。该结合阈值内置于每个矩阵中,描绘了得分最高的肽之间假定的结合物的范围[39]。
IEDB分析资源数据库自2011年起就使用NetMHCpan作为预测方法。该方法可对任何肽类-MHC I类相互作用进行亲和力的定量预测,涵盖人类的HLA-A和HLA-B以及黑猩猩,猕猴,大猩猩, 牛,猪和老鼠。 这是包括这种生物的少数几个数据库之一[37]。
nHLAPred是另一个用于预测67个MHC等位基因MHCI结合肽的综合工具。 等位基因的预测基于人工神经网络(ANN)和定量矩阵(QM)。 通过使用蛋白酶体基质,将预测的MHC结合物过滤到潜在的CTL表位。 尽管该服务器提供了两个选项(Compred和ANNPred),但范围最广的是Compred, 基于人工神经网络和定量矩阵的混合方法[40]。
NetMHC服务器使用ANN预测肽与许多不同的HLA等位基因的结合。 人工神经网络已针对代表所有12种HLA-A和-B超型的78种不同人类MHC(HLA)等位基因进行了训练。 此外,还有41种动物的预测,如猴子,牛,猪和小鼠等位基因[38]。
基于内核的等位基因间肽结合预测系统(KISS),使用支持向量机(SVM)多任务内核来预测等位基因肽是否会结合64个等位基因的MHC-1分子,从而利用等位基因之间的可用训练信息, 提高了其准确性,尤其是对于很少有已知表位的等位基因。 在包含SYFPEITHI,MHCBN,LANL和IEDB数据库中已知表位的数据库上训练预测变量。
尽管还有其他服务器可用于识别MHC-1结合预测因子,但上述服务器在等位基因覆盖和人类以外其他生物体等位基因的识别方面是最完整的,但是可提供更详细的MHC-1结合预测因子列表 表1列出了在线内容。

TAP是与MHC I类限制性抗原加工相关的转运蛋白。 TAP是异源二聚体转运蛋白,属于ABC转运蛋白家族,它利用ATP水解提供的能量使肽段穿过内质网膜转运。转运蛋白由两种名为TAP-1和TAP-2的蛋白质组成。该转运肽的子集将结合MHC I类分子。这些MHC肽复合物易位在抗原呈递细胞(APC)的表面,随后可能引发T细胞免疫反应[41],[42],[43]。可用的TAP绑定预测软件包括TAPPred,EpiJen和WAPP(表2)。 TAPPred是一种在线工具,可预测肽对TAP转运蛋白的结合亲和力。 TAP结合肽的预测对于鉴定MHC-1限制性T细胞表位至关重要。该预测基于级联SVM,使用氨基酸的序列和特性[44],[45]。 EpiJen服务器不仅提供TAP结合预测,还提供蛋白酶体截止(proteasome cut off)。 EpiJen使用加成法,该方法假定每个取代基对生物活性起加成和独立的作用。他们的加成法考虑了特定氨基酸和结合位点之间的相互作用[46]。
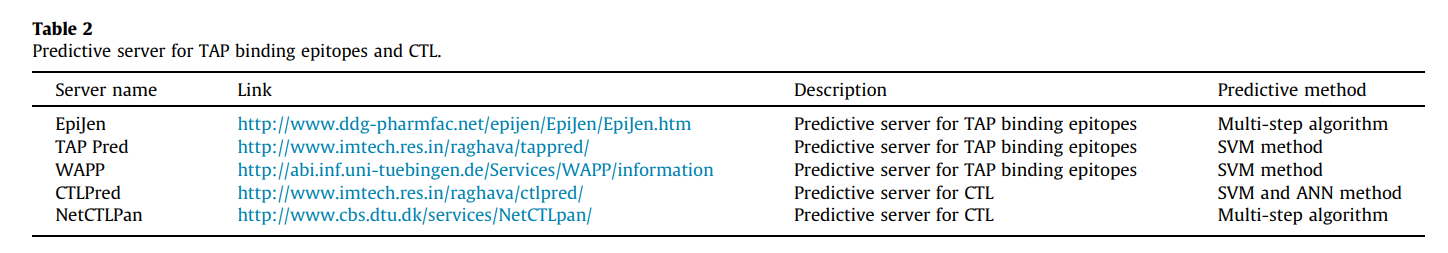
完整抗原处理途径(WAPP)服务器包括用于预测蛋白体切割,TAP转运和MHC肽结合的工具。该服务器针对这三个方面提供了集成的预测。蛋白酶体切割的预测是基于对烯醇酶和Pri病毒蛋白进行的实验。实验切割位点周围的序列用于构建权重矩阵,而SVM的回归形式则用于预测TAP亲和力[47]。 MHC I类途径的预测可以通过蛋白酶体切割,TAP转运效率和MHC I类结合亲和力的预测得到改善[48],[49]。
然而,尽管对I类MHC的预测已取得了良好的表现,但在预测MHC-II结合表位方面仍然取得了有限的成功[50],[51]。 MHC-II结合表位的预测准确性低是由于以下几个因素造成的,包括:
- 训练数据不足或质量低,
- 难以在用于训练的较长肽中鉴定9-mer结合核心
- 缺乏考虑侧翼残基的影响
- MHC-II分子结合槽的相对介电常数限制了结合严格性[29],[50]。
ProPred1是使用定量矩阵预测抗原序列中MHC II类结合区域的服务器。服务器将协助找到混杂的结合区域,这对于选择覆盖51个等位基因的候选疫苗很有用[52]。 SVMHC服务器可以预测MHC I类和MHC II类结合肽,但是最广泛的覆盖范围是MHC-II(51个等位基因)。该软件中显示的图形输出还允许简单识别混杂表位。 SVMHC使用由TEPITOPE软件开发的矩阵[47]。
MHC2Pred是用于预测混杂MHC II类粘合剂的基于SVM的方法。基于支持向量机的方法对42个等位基因的平均准确度约为80%。由于数据集较小,该方法对于少数等位基因的性能较差。该服务器将用于细胞免疫学,疫苗设计,免疫诊断,免疫治疗以及自身免疫易感性的分子理解[52]。这最后三台服务器以及上述RANKPEP是最完整的服务器,具有最广泛的等位基因覆盖范围,可预测表位与MHC-II的结合,并且还具有友好的界面。表1描述了预测MHC II类结合表位的其他资源。
还有另一种直接的CTL表位预测方法,这对亚单位疫苗设计至关重要。 这些服务器的示例是CTLPred和NetCTLPan。 CTLPred使用T细胞表位模式代替MHC结合物。 该方法基于诸如ANN和SVM之类的技术。 这些方法还允许基于这两种方法进行共识和组合预测[53]。
NetCTLpan服务器可以预测蛋白质序列中的CTL表位,并使用人工神经网络。 该方法已更新,包括IMGT / HLA和IPD-MHC数据库(适用于非人类灵长类和猪)中最新的MHC等位基因。 在此软件中可以预测8-11mer肽段,因为大多数HLA分子对结合9mers有强烈的偏爱[表2和48]。
尽管某些方法根据其特征和局限性比其他方法更适合,但上述表位发现工具可以轻松应用于大多数病原体。 但是,应该记住,这一预测是潜在功能的指标,而不是功能分配的标准。 因此,应将这类计算机模拟分析与其他证据(包括实验数据)结合起来以分配功能。
四、预测潜在B细胞结合表位的生物信息学工具
B细胞表位被其天然结构中的B细胞受体或抗体识别。 连续B细胞表位预测与T细胞表位预测非常相似,后者主要基于氨基酸特性,例如亲水性,电荷,暴露的表面积和二级结构。 不连续的B细胞表位预测需要抗原的3D结构[54],[55],[56]。 迄今为止,网上有一些特定的资源可以预测连续或不连续的B细胞表位(表3)。
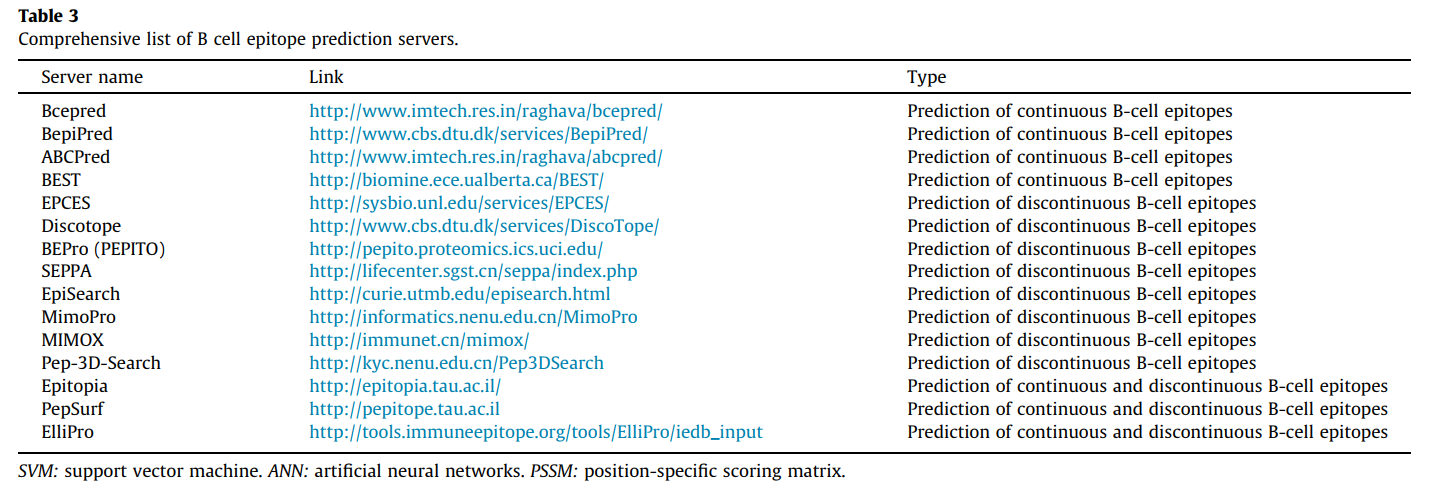
为了预测线性B细胞表位,Bcepred工具基于非冗余数据集上的理化特性,例如亲水性,柔韧性,极性和裸露表面。 该数据集由从Bcipep数据库获得的1029个B细胞表位,和从Swiss-Prot数据库中随机获得的相等数量的非表位组成。 基于这些属性的模型的预测准确度从52.92%到57.53%不等[57]。 基于神经网络的ABCpred服务器的估计准确性为65.93%[54]。 另一台名为BepiPred的服务器使用隐马尔可夫模型和倾向标度方法的组合预测线性B细胞表位的位置[58]。 上面提到的服务器易于使用且结构合理。
在用于预测不连续B细胞表位的工具中,我们可以提到DiscoTope,它使用蛋白质的三维结构来确定表面可及性和新的表位倾向性氨基酸评分。 最终得分是通过将空间接近度中的残基倾向得分和接触数相结合来计算的。 该服务器还可以预测多链复合物中的表位[59]。 该工具与BEpro(以前称为PEPITO)和SEPPA(蛋白质抗原的空间表位预测)一起需要3-D结构作为输入,特别是PDB格式[60]。 使用SEPPA,将根据来自其邻近残基的信息为查询蛋白中的每个残基评分。 较高的分数对应于残基参与表位的可能性较高[61]。
该领域最完整的工具之一是ElliPro。 该服务器根据蛋白质抗原的3-D结构预测线性和不连续的表位。 ElliPro将每个预测的表位与一个得分相关联,该得分定义为PI(突出指数,Protrusion Index)值。 与前面提到的数据库相比,在ElliPro中输入的是蛋白质序列。 通过基于用户选择的结构模板的同源性建模,可以预测输入蛋白质序列的3-D结构。 之后,将基于预测的蛋白质结构计算线性和不连续的表位。 表3包括一些其他生物信息学工具,可预测连续和不连续的B细胞表位。
所有这些综合工具为开发新疫苗特别是旨在引起体液反应的疫苗提供了机会。
五、针对高变病毒的基于新兴肽疫苗的生物信息学策略
历史上,大多数已知的成功疫苗都是凭经验开发的。 但是,高度复杂的病毒(如具有高度遗传和抗原多样性特征的HIV和流感病毒)的出现,阻碍了使用传统方法开发有效的,广泛覆盖的疫苗。 这些病毒病原体的快速出现突显了对改进和加速开发和生产疫苗的过程的需求,可以通过上述方法解决上述需求,该方法允许采用基于计算机技术的快速方法来配制疫苗候选物。
本节简要讨论了针对人类免疫缺陷病毒(HIV)和流感病毒开发的一些方法,以举例说明如何使用生物信息工具在高变病毒的情况下成功实现候选疫苗设计。
5.1 艾滋病病毒
考虑到其在全球的流行重要性,成功的抗HIV疫苗是当务之急。 随着HIV击败旨在中和它的免疫系统,这种发展对研究人员构成了重大挑战。 另外,遗传物质可以保持休眠状态,从而逃脱了免疫系统[62]。 尽管抗逆转录病毒药物可以控制许多患者的HIV / AIDS进展,但它们只能成功地减少病毒载量而不能完全消除病毒[63]。 因此,开发有效的HIV-1疫苗代表了控制大流行HIV的最佳解决方案。
Diaz-Mitoma及其同事[64]已完成了一项高级概念验证,他们开发了基于Variosite的HIV-1疫苗。 该候选疫苗包含176个肽库,分别代表gp120包膜的五个高变表位和gag的两个可变表位。 针对非人类灵长类动物模型中的一组异源HIV-1亚型,测试了该多价疫苗的效力和覆盖范围。 检测到针对HIV-1亚型A–F的特异性CD8 + T细胞免疫反应,这非常显着,因为亚型中的HIV-1序列相差高达20%,而亚型之间的HIV-1序列相差高达35%。 在免疫的动物中还表征了结合抗体的效价和中和活性,观察到所有动物中针对变体gp120蛋白的IgG抗体效价的水平很高。 此外,六分之三的免疫猕猴对两种原发性HIV-1具有中和活性[64]。
另一方面,黄等[65]报道了一种预测构象B细胞表位的方法。为了确定特定肽段中的某些区域是否可以构成B细胞表位,将噬菌体展示的随机肽库用作鉴定拟态的有力工具。通过以与天然表位相似的模式结合给定的单克隆抗体(mAb)进行选择。这些模拟表位可以被认为是功能性表位模拟物。这种方法不仅可以预测线性表位,还可以预测构象表位,因此,该方法代表了该领域的重要策略。此方法称为Pep-3D-Search,它依赖于特定抗原的3D结构和一组模拟表位(或衍生自该模拟表位的基序序列),并可以两种方式使用:模拟表位或主题。为了评估从一组模拟表位预测表位的能力,将晶体学定义的10个表位与Pep-3D-Search的预测结果进行了比较。与其他可用的预测算法相比,Pep-3D-Search显示出可比的Matthews相关系数(MCC),特异性和精确度,并且可以提供新颖,合理的结果。另一方面,作者验证了Pep-3D搜索将基序序列与3-D结构比对以预测表位的能力。分析了六个测试用例,其中包括三种HIV蛋白质,证明其性能优于其他类似程序。另外,该程序能够快速定位由更长的同位素模仿的表位区域。尽管此有前途的工具提供了一种强大的方法来定位模拟表位模拟的表面区域,但有必要评估这些鉴定出的表位的免疫保护能力。
在抗体-抗原复合物的原子级结构的背景下,在许多情况下确定结构可能是不切实际的。最近,Georgiev及其同事[66]描述了一种有效的计算方法,以预测残基水平的抗体特异性HIV-1包膜(Env)表位。该方法包括评估代表抗原的一系列不同病毒株的中和力数据。通过结合来自抗原未结合结构的信息可以提高准确性。具体而言,在包含181种不同病毒株的中和板中评估了19种HIV-1 Env抗体,并在分析中考虑了可用的抗体-抗原复合物结构。预测效率比随机预测高8倍。此外,当用于前瞻性预测两种HIV-1抗体8ANC131和8ANC195的表位残基时,该工具可进行成功的预测,并通过实验验证。该程序固有地适用于表现出序列多样性的抗原,显示出与表位序列保守性成反比的准确性。这些见解表明,如何将源自中和小组和未结合抗原结构的数据用于抗体表位的残基水平预测,代表了一种产生针对高变病原体的有效疫苗的重要方法[66]。
表4集中了基于反向疫苗学方法的这些努力的代表性观点,并反映了在该重点下克服上述障碍的可行性。 因此,生物信息学工具提供了可预测疫苗靶标的强大资源。 表4中提供了其他情况。

5.2 流感 Influenza
流感是一种具有高度传染性的空中呼吸道感染,伴有严重的疾病负担。 新的流感亚型会定期出现,在人群中不存在免疫力,因此可能导致全球大流行。 新型流感亚型的爆发例证了这种病原体如何发展大流行[67]。 由于快速的突变率,病毒的可变和发散特性以及极长的疫苗开发流程,寻找一种有效的,广泛覆盖的流感疫苗是一项令人沮丧的任务。
一些研究表明,使用免疫生物信息学预测因子可以更快,更全面地筛选表位以设计候选疫苗(参见表5)。 2007年,Wang等 [69]通过使用SYFPEITHI软件,鉴定了十种新型流感抗原表位,并确认了三种先前已知的仅限于细胞毒性T细胞(CTL)的抗原。 这些表位保存在高致病性H5N1流感病毒的不同分离株中,所有这些也存在于新兴的禽流感分离株中。 通过ELISPOT测定法评估了预测的肽的免疫原性。 这些表位可用于检测患者的流感特异性CTL反应。 此外,他们的研究结果对疫苗的合理设计具有重要的意义,因为疫苗的个体表位或融合为多表位的表位适用于所有种族。

Cheung等人采用类似的方法。 [70]使用SYFPEITHI软件将甲型流感病毒(H5N1株)作为目标,实现了九种潜在免疫原性肽的鉴定。 进行了体外测定以确定这些肽的免疫原性潜力。 他们的发现凸显了表位AMDSNTLEL和QGRGVFEL的潜力。 这种新颖的细胞毒性T细胞表位构成了人类H5N1疫苗开发的相关信息。
另一方面,De Groot等使用EpiMatrix(T细胞表位预测和比较工具)。 [30]比较了甲型流感病毒的三种血凝素(HA)和神经氨酸酶(NA)蛋白的序列。在计算机分析中发现,HA序列中包含16个混杂的辅助T细胞表位,其中9个在2008-2009年流感疫苗株中100%保守。在同一小组的后续研究中,使用来自人类供体的外周血单核细胞对选定的表位进行了生物学研究。 IFN-γELISPOT和CD4 + T细胞刺激试验可以评估原代和升压后T细胞反应,观察计算机预测与观察到的反应之间的相关性,对CD4 + T细胞表位的准确预测为80-90%。这些发现反映了这种计算工具的鲁棒性[71],对抗高变病原体的新疫苗的配制具有影响。表5显示了在开发新的流感疫苗时应用的其他基于生物信息学的方法,这些方法合在一起证明,可以通过计算机基因组分析快速识别潜在的表位;然后确认进行实验评估。
六、展望
生物信息学工具使人们能够选择潜在的抗原决定簇,而无需承担培养感兴趣的病原体所涉及的风险。 与传统的疫苗学技术相比,这种方法具有巨大的优势,包括更快的产量和更低的成本。 “组学”技术在该领域的应用也彻底改变了识别潜在候选疫苗的方式。 蛋白质组学和转录组学已被用作基因组学的补充方法,通常在鉴定宿主与病原体之间的相互作用过程中对表面蛋白的鉴定更为有用。
尽管有许多表位预测方法可用,但仍需要在标准基准数据集上开发对不同方法的系统评估。开展针对表位预测的关键技术评估确实将使该领域受益。已经提出,将使用计算方法从先前通过实验筛选的查询蛋白中进行盲的从头表位预测[72],[73]。由于许多方面,包括以下几个方面,比较不同方法仍然是一项复杂的任务:
- 数据集和预测方法的文档不足,
- 用于评估方法的基准数据集不可用,
- 实现方法(iv)缺乏统一的输出格式,这使合并多个服务器的结果以获得共识预测的过程变得复杂[74],[75]。
因此,有必要开发标准化的数据表示形式。这将能够对标准化基准数据集上的不同预测方法进行评估,以便比较这些方法并开发结合多种预测工具的预测的元服务器[55]。
例如,Epitopes Toolkit(EpiT)是开发表位预测方法的平台设计。这使其他研究人员可以在自己的计算机上使用已开发的预测器,在其他数据集上重建预测器,或将预测器与其他预测器组合以获得定制的混合或共识预测器。 EpiT包含两个组件:(a)模型构建器,一个用于构建和评估抗原决定簇预测因子并以二进制格式序列化这些模型的应用程序; (b)预测器,用于将模型应用于测试数据的应用程序。尽管EpiT是为开发表位预测工具而设计的,但EpiT的某些组件可用于不同的序列分类任务。此外,EpiT中的某些数据预处理器可以应用于亚细胞定位,以及其他蛋白质序列分类[74],[75],[76]。该平台是用Java实现的,可以从项目网站 http://ailab.cs.iastate.edu/epit 免费下载。该网站还为表位预测工具的开发人员和EpiT用户提供了丰富的资源。有用的资源包括:Epit文档,扩展的表位预测变量存储库和表位数据集存储库[74]。
其他限制与使用数据集训练表位预测因子有关,其中一些表位残基被错误地标记为非表位残基,因此预测因子在此类数据集上的表现往往会夸大假阳性的数量。 许多作者[77],[78],[79],[80]试图通过使用从SwissProt蛋白质序列随机样本中提取的非表位残基来改善这一限制[81]。
另外,为了改善不同表位预测方法的局限性,加速分析方法的发展,进行比较分析是必不可少的。 已证明对表位预测方法的关键评估在其他领域是合适的[82]。 使用大型的,非冗余的以及实验性良好的数据集可以帮助提高基于交叉验证的抗原决定簇预测因子性能评估的准确性。
上面描述的所有生物信息学工具都提供了完整的表位列表,以设计候选疫苗。 如何决定从大量抗原决定簇中选择哪个进行临床试验仍然是一个挑战。 尽管基于基因组的方法极大地加速了疫苗靶标的鉴定,但疫苗学家仍然必须经历繁琐而缓慢的验证过程。
因此,使用这些工具鉴定的新的基于候选抗原决定簇的疫苗必须接受现有的标准体内验证研究(例如动物保护实验),就像使用常规方法鉴定的疫苗抗原一样。
不同的数据库可用于预测T细胞和B细胞表位。然而,在预测B细胞表位的工具中,仍需要进一步发展,因为理想的工具应考虑数据集中的成分偏差[83],[84]。优先识别的抗原表位的实际预测关键取决于蛋白质组成。大多数可用的表位预测方法都是基于氨基酸特性,包括亲水性[85],[86],溶剂可及性[84],二级结构[87],柔韧性[88]和抗原性[83]。在2005年,Blythe和Flower证明,使用单尺度氨基酸倾向图谱不足以可靠地预测表位的位置,而在2007年,Greenbaum等人(2002年)提出了这一点。结果表明,结合使用多个氨基酸倾向量表和机器学习算法可以提高预测准确性。这样的研究对表位的预测显然具有免疫学意义[55],[89]。
电脑抗原表位预测的性能不佳可以在许多报告中找到。 例如,Blythe和Flower检查了预测性B细胞表位与来自50种蛋白质的表位位置之间的相关性,以进行多克隆反应。 他们发现,单尺度氨基酸倾向图不能用于可靠地预测表位的位置,因为生成的序列图和已知线性表位的位置之间不存在相关性。 几种算法的适当组合以及对生成信息的严格评估,对于成功选择抗原表位至关重要。 因此,疫苗学家应意识到以下事实:产出高度依赖于工具选择的标准。 这一事实凸显了任务的复杂性,并激发了开发更复杂,更具体的工具以提高性能的效果[55]。
在Wang等人的报告中。 MHC-II预测工具的性能不如I类预测所指出的那样,这可能是由于MHC II类需要高度特定的匹配这一事实。 最初使用晶体结构中显示的9-mer肽核评估了预测能力。 从评估的八种算法中,观察到除PROPRED和SYFPEITHI之外的较差性能,这反映了识别正确的结合核心的困难。 为了改善这些局限性,通过将这两种II类结合预测方法相结合,开发了一种共识预测工具,与14种候选结合MHCII中的10种相比,与单个输出相比,观察到的性能得到了改善[90]。
Resende等人发表了一个有趣的例子。 他报告了对五种表位预测算法(NetCTL,NetMHC,BepiPred和AAP12)和三种亚细胞定位预测算法(以锥虫为靶标基因组)的性能的评估。 算法之间的比较是基于AUC(ROC曲线下的面积)值进行的,该值表示随机选择的阳性表位得分高于随机选择的阴性表位的概率。 AUC数据表明,NetCTL和Net MHC(分别为0.66和0.60)性能之间仅存在很小的差异。 考虑到MHC-I预测方法已达到0.95-0.99的AUC值,因此将NetCTL和Net MHC确定为低性能方法。 另一方面,对B细胞表位算法(BepiPred和AAP12)的AUC值的评估显示,其性能比以前的算法要差[91],[92]。
因此,可以指出不同的预测模型在性能上会有所不同,因此应针对特定的目标生物评估一套工具。然后,预期该组合预测方法将在疫苗开发领域中提供扎实的进展。
一旦完成对目标表位的选择,就可以基于合成的肽混合物设计疫苗制剂,所述混合物可以包括佐剂序列,例如Th表位,以增强免疫原性[综述见93]。然而,合成肽对于临床应用而言是昂贵的。一个有吸引力的选择是设计带有目标表位集合的重组蛋白[94]。在嵌合蛋白中起重要作用的其他序列是连接子和佐剂序列,它们是促进折叠以实现适当的表位展示的间隔子。 Thomas和Luxon [95]和Cozzi等人发表了关于疫苗嵌合蛋白设计的出色综述。 [96]。
我们的小组已经生产并评估了多表位蛋白,作为抗传染病的候选疫苗。 这些候选物在小鼠模型中的免疫原性已被证明是有希望的,因为诱导了针对不同靶标的体液反应[97],[98],[99]。 这些设计包括主要由传统的免疫原性试验(例如抗原决定簇作图以及血清学和功能测定)描述的抗原决定簇。 可以预见,本综述中概述的生物信息学工具可能会导致针对这些病原体的创新嵌合设计。
这些技术还将在生物医学领域取得重要进展的特定情况包括艾滋病毒/艾滋病和流感。尽管存在全球艾滋病毒/艾滋病的流行病,但经过数十年的重点研究,寻找一种有效的疫苗仍然很困难。然而,RV144泰国试验产生了新的希望,其中确定了适度的保护潜力[100]。与观察到的免疫保护作用相关的表位正在被鉴定,并且基于这些表位的进一步疫苗开发有望提供新的有前途的候选物[101]。理想的方法是开发由选定的保护性抗原决定簇组成的疫苗,因此是抗击艾滋病毒/艾滋病的机会领域。
就流行性感冒而言,尽管有保护性疫苗可用,但它们涵盖了某些变种,但有防范新变种的风险,因此应定期生产新疫苗以对抗新出现的变种。通用疫苗是提供针对多种菌株的广谱免疫力的理想方法。在迄今为止研究的候选对象中,可以提到甲型流感病毒M2蛋白的高度保守的胞外域和茎域[102]。还测试了使用核蛋白和M1蛋白的基于T细胞表位的疫苗。包含这些有前途的表位的多表位设计预示着新的潜在流感疫苗,作为针对这种相关的高变病原体的普遍干预的模型[103],[104]。
总而言之,在开发可用于疫苗合理设计的工具方面进行了广泛的研究,这是相当可观的,但计算机模拟分析的进一步改进以及实验评估对于在疫苗开发领域取得进展,最终将这些技术衍生的新疫苗推向市场至关重要,特别是对于高度可变的病毒病原体[105],[106]。
参考资料
- Journal of Biomedical Informatics Volume 53, February 2015, Pages 405-414 。https://www.sciencedirect.com/science/article/pii/S1532046414002330
