【4.1.1】淋巴细胞受体结构建模--lyra
官网:http://tools.iedb.org/lyra/
B细胞和T细胞受体的准确结构模型是获得深入了解免疫机制以及开发新药和疗法的基础。 LYRA(淋巴细胞受体自动建模)Web服务器( http://www.cbs.dtu.dk/services/LYRA/ )实现了从其氨基开始构建B细胞和T细胞受体结构模型的完整自动化方法单独的酸序列。该网络服务器可免费使用,并且对于非专业人员而言易于使用。提交后,LYRA会使用临时配置文件自动生成比对,预测每个高变环的结构类别,以自动方式选择最佳模板,并在几分钟内提供可在线下载或检查的完整3D模型。有经验的用户可以根据案例的具体信息手动选择或排除模板结构。 LYRA基于规范的结构方法,在过去的30年中已成功用于生成高精度的抗体模型,在我们的基准测试中,该方法在TCR建模方面证明也取得了类似的良好结果,基准的平均RMSD精度为对于B细胞和T细胞受体,分别为1.29和1.48Å。据我们所知,LYRA是第一台用于预测TCR结构的自动化服务器。
一、前言
免疫系统有能力针对极端分子和外源性(病原体,毒素)或自体源性(肿瘤,代谢副产物)的危险分子并与之抗争。这种非同寻常的防御机制中的两个关键分子是T细胞受体(TCR)和B细胞受体(BCR,抗体或免疫球蛋白),它们之间的相互作用确保了精确而受控的免疫反应。为此,在高等生物的所有细胞类型中,特异性针对其产生的淋巴细胞具有通过基因组重组控制其基因组序列的能力,可以根据其能力对其进行正选择或负选择识别自身和非自身分子,并在B细胞的情况下,通过称为“亲和力成熟”的过程进一步改变其基因组含量。所有这些事件最终使生物体产生了巨大而又高度受控的不同淋巴细胞受体库(1)。
抗体识别血液或粘膜组织中存在的潜在有害分子(抗原),并且是抵抗感染的第一个障碍。研究和预测其结构的能力对于我们对免疫系统,病原性和自身免疫性疾病的理解(2-4)以及对新疗法和新药物的开发(5)至关重要。即使一个人产生的抗体多样性比所有其他人类蛋白质的多样性都更大(6),我们仍可以以极高的准确性预测其结构(7,8)。这种非凡的能力源于Chothia和Lesk在30年前所做的一项基本发现(9-11):尽管T细胞和B细胞受体的序列变异性很大,尤其是在它们的抗原结合位点(ABS)上,但这确实对其保守的主链构象没有可比的影响。组成ABS(抗体结合位点)的六个CDR(互补决定区)环只能采用有限量的通常被特定序列特征识别的命名为规范结构的构象。规范结构模型已被证明对T细胞和B细胞受体均有效,但是直到现在,它仍被开发和实现为仅用于B细胞受体(BCR)分子的自动建模工具。这种偏见的原因在于大量具有可用的已解决结构的抗体,以及在制药和工业应用中大量使用抗体。然而,在最近几年中,T细胞受体(TCR)在开发用于治疗癌症(12,13),过敏和自身免疫性疾病(14,15)的疫苗和疗法及其精确模型方面获得了越来越多的关注。结构已成为该领域发展的基本步骤。
在这里,我们介绍LYRA(淋巴细胞受体自动建模),这是一种用于对B细胞和T细胞受体进行自动建模的Web服务器。 它基于规范的结构方法,可以在几分钟内轻松生成极其可靠的淋巴细胞受体模型。
二、材料和方法
2.1 建模管道 Modeling pipeline
使用内部生成的序列配置文件扫描输入序列。 对于每个输入序列,使用最佳得分的资料来推断受体和链的类型以及正确的比对。
- 如果LYRA能够识别并正确排列给定淋巴细胞受体的两条链,则管道会继续从精选模板的数据库中自动选择最佳框架模板,
- 最后自动选择需要移植的CDR模板。
- 然后合并模板,并重新打包侧链以生成最终模型。
总体建模过程平均不到一分钟(5 s排队时间,30 s计算时间)。 每个建模步骤的详细说明如下。
2.2 模板数据库
使用Pisces Web服务器(17)检索IMGT / 3Ds tructure-DB(16)中存在的所有BCR和TCR结构的PDB code,并删除所有冗余结构。具有至少一个非冗余链的任何完整分子以及非冗余非配对链都保留在数据库中。生成的TCR结构数据库由105个成对的链,两个单独的α链和九个单独的β链结构组成。 BCR结构数据库由846条复杂链对,10条独立的重链,5条独立的kappa链和1条独立的lambda链组成。使用临时序列配置文件对所有结构进行了比对和重新编号(请参阅下一段)。模板数据库每月自动更新。检查存储在PDB数据库( http://www.rcsb.org/pdb/ )中的所有新颖的已解析结构,并将与当前版本数据库中的任何分子均非冗余的结构添加到该结构中(请参阅补充资料)详细资料。
2.3 序列资料 Sequence profiles
LYRA使用BCR重链,λ和κ链以及TCRα和β链的序列图谱来识别和比对淋巴细胞受体序列。 BCR HMM是根据作者先前开发的协议生成的(18)。最终的HMM分别包含针对lambda,kappa和重链的5462、12930和36895序列,这些序列根据Kabat-Chothia编号方法进行了排列,并带有Abhinandan和Martin(19)所述的其他插入物。
为了生成TCRα和β链的HMM分布图,我们使用t-coffee软件版本11.0010(20)的3DCoffee / TMalign模式生成了上一步中检索到的212 α链和221 β链结构的多重结构比对(不包括冗余减少)。 然后将从这些结构比对中提取的多序列比对(MSA)用作HMMER版本3.1(21)(hmmbuild,默认设置)的输入,以创建种子HMM配置文件。然后从IMGT / LIGM-DB(22)下载有关α和β链的其他核酸序列,将其翻译为氨基酸序列,并使用种子HMM图谱进行比对。所有长于100个残基且E值低于10-50(501α链和599β链序列)的命中均添加到相应的MSA中。与我们在Kabat-Chothia比对中观察到的一致(10),然后将MSA的CDR区中的插入右对齐,以便在每个CDR比对的C端区域中有一个重新进入的位置。框架区域中的插入未编辑。所得的比对用于生成最终的HMM。
2.4 规范结构 Canonical structures
其他人和我们先前已经定义了BCR模型的规范结构(CSs)和相应规则(10,11,23–28)。为了生成用于TCR CDR循环的CS类和预测规则,我们采用了North等人的方法。 (29)。 CDR环按长度和相同长度的距离来分组,相同长度的每对环之间的距离计算为两个环中每个残基的phi和psi角的距离之和,该距离定义为D(θ1, θ2)= 2(1-cos(θ1-θ2))。 CS是通过将Python软件包scikit-learn(30)中的亲和力传播聚类算法应用于针对每种链类型,CDR和环长度而获得的距离矩阵来获得的。补充表S1和S2分别报告了TCRα和β链的所得CS。继Chailyan等(23,24),我们训练了一种基于序列的预测方法,该方法在给出TCR链的序列的情况下,可以预测其CDR的CS。有关TCR规范结构(补充表S1和S2),CS预测程序及其准确性(补充表S3-S5和补充图S1-S3)的更多详细信息,请参见补充材料。
2.5 模板选择 Template selection
如前所示,模板选择步骤对于模型的准确性至关重要,这取决于一方面更高的序列相似性和另一方面需要将来自不同模板的区域合并在一起的需要(18)。为此,我们结合使用了不同的分数和选择程序。给定对齐的目标序列,我们为每个模板计算五个分数。前四个得分是相似性得分,使用BLOSUM62相似性矩阵,一个完整序列,三个CDR中的每一个。最后,有一个组合分数,计算为完整序列分数与模板CDR得分的总和,该模板CDR匹配相应的目标CDR规范结构。
为了使框架模板具有相同的晶体结构,从而避免将不同的链包装在一起而可能引起的错误或冲突,使用了以下管线:对于每个目标链,组合得分最高且序列相同的20个模板列出目标至少为60%的目标,并选择来自相同晶体结构且具有最高整体序列同一性的模板。如果找不到相同晶体结构的模板对,则程序将退回以为每个单独的链选择具有最高组合环得分的框架模板,从而最大程度减少最终要嫁接到模型中的CDR环的数量。 。
接下来,扫描每个查询链中的每个循环(β CDR3除外)。如果所选框架模板中循环的规范结构与查询序列的计算出的规范结构匹配,则保持该循环不变,否则,将选择具有相同规范结构和最高CDR特定得分的循环作为模板。在数据库中找不到这样的循环的情况下(模板黑名单,没有预测的规范结构),将选择与模板循环长度相同且CDR得分最高的模板循环。此选择方法始终用于无法识别清晰CS的β CDR3环。
2.6 模型组装
最终的模型组装包括三个阶段:
- 两个链的包装
- 环接枝
- 侧链建模。
如果为两条链选择的框架模板均来自不同的晶体结构,则根据确定界面堆积的位置,使用一组界面堆积残基构建“伪序列”。选择一对具有相同晶体结构且基于伪序列具有最高BLOSUM62相似性的模板,并通过重叠属于该界面的所有残基的目标残基和模板残基对两个模板链进行建模(18,23)。
通过将两个残基的骨架原子叠加在环的N末端残基之前和两个残基紧接在环的C末端之后,可以移植非框架模板的模板环。
然后,使用Scwrl4软件重新包装CDR3残基的侧链以及在靶标和模板之间不保守的每个残基的侧链(31)。
最后,为了消除碰撞和不良的几何形状,使用ENCAD程序对该模型进行500步能量最小化(32)。
三、网页界面
LYRA( http://www.cbs.dtu.dk/services/LYRA/ )提供了易于使用的界面来输入氨基酸序列数据,结果页面包含模型的图形化和可下载输出以及高级面向经验丰富的用户的建模选项,以及包括示例序列和输出的详细帮助页面。在下文中,我们将更详细地描述LYRA Web服务器的主要元素。可以在LYRA帮助页面上找到更多信息和更详细的指南。
3.1 输入页面
LYRA输入页面的主要输入包含TCR或BCR两条链的氨基酸序列。每个序列都应以原始格式(即没有标题或特殊字符)复制到两个文本区域之一,分别标记为“链1蛋白序列”和“链2蛋白序列”。 “ PDB黑名单”输入区域(可选)允许用户输入以逗号分隔的任何PDB结构列表,这些列表不应用作模板,因此应在建模管道中丢弃。标有“ TCR”和“ BCR”的两个按钮将分别用示例BCR或TCR序列加载文本区域,覆盖已复制在输入区域中的任何序列。按下“提交”后,用户将被重定向到一个队列页面,该页面将重新加载直到作业完成。用户可以选择在此页面上提交他/她的电子邮件地址,并接收带有该作业链接的邮件。该字段不是必填字段,只需在页面上加书签即可在以后访问结果。
3.2 输出页面
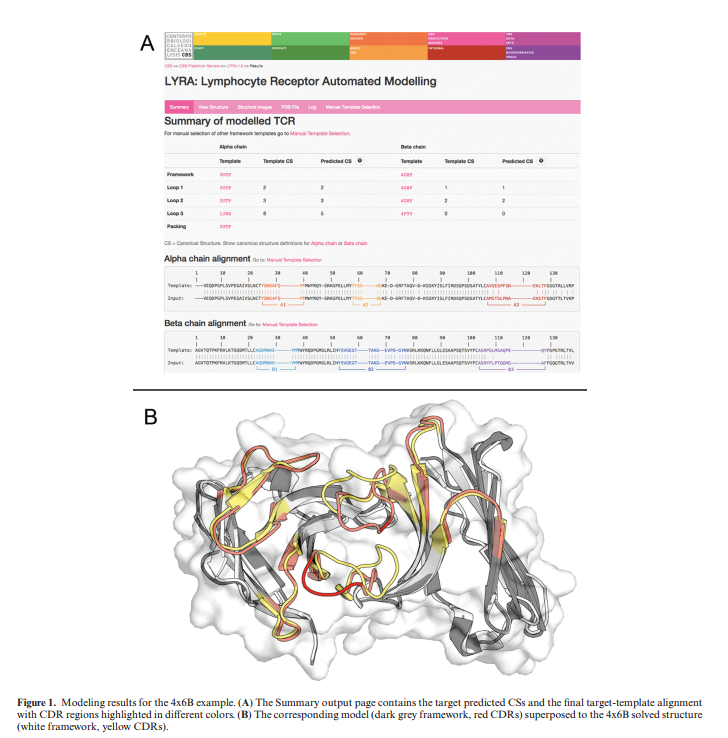
LYRA输出页面包含一个链接到多个选项卡的标题栏。 “摘要”标签显示了有关建模过程的详细信息:两个链均报告了用于建模框架的模板和每个链的CDR环。对于每个CDR环,显示了靶标和相应模板的CS。通过单击相应链类型附近带有问号按钮的按钮,可以显示CS的完整列表。用户可以通过“视图结构”选项卡中的JSMol应用程序以交互方式可视化模型,也可以使用“结构图像”选项卡中使用PyMOL软件生成的高分辨率渲染来可视化模型。在这两种情况下,CDR回路都使用不同的颜色突出显示。具有完整节能模型的PDB文件可以在“ PDB文件”标签中在线检查,也可以通过单击“下载PDB文件”按钮进行下载。值得注意的是,LYRA生成的pdb文件的标题部分包含在输出页面上找到的大多数建模详细信息。 “日志”标签显示有关作业输入,输出和可能的错误的技术信息。
在“手动模板选择”中,用户可以通过单击页面中列出的任何模板来覆盖自动选择框架模板。对于每个模板,列出了所有相关信息,例如其CDR环的规范结构,相对于靶序列的序列同一性和Blosum评分。在页面底部可见目标序列与当前所选模板的比对。通过单击此选项卡中的“提交”按钮,将使用刚选择的框架模板和CDR循环的自动选择的模板来启动新作业。单击标题栏中标有“更多”的最右边的按钮,可以下载其他文件。这些文件包括JSON和csv格式的输入分子和建模参数的完整摘要,包含与’结构图像’选项卡中显示的图片相似颜色的模型的PyMOL脚本,‘Pre-SCWRL’pdb,其中仅存在模板中保守的主链原子和侧链,以及一个非最小化的完整模型,可用作用户选择的侧链建模和能量最小化软件的输入
3.3 帮助页面
LYRA服务器提供了一个全面的帮助页面,可以从输入和输出页面的标题栏中访问该页面。帮助页面包含LYRA服务器及其用法的简短描述,建模管道的概述,每个淋巴细胞受体链中框架和CDR区域的详细定义,指向每种链类型中具有CS定义的表的链接以及服务器输出的描述。
四、评估和案例研究
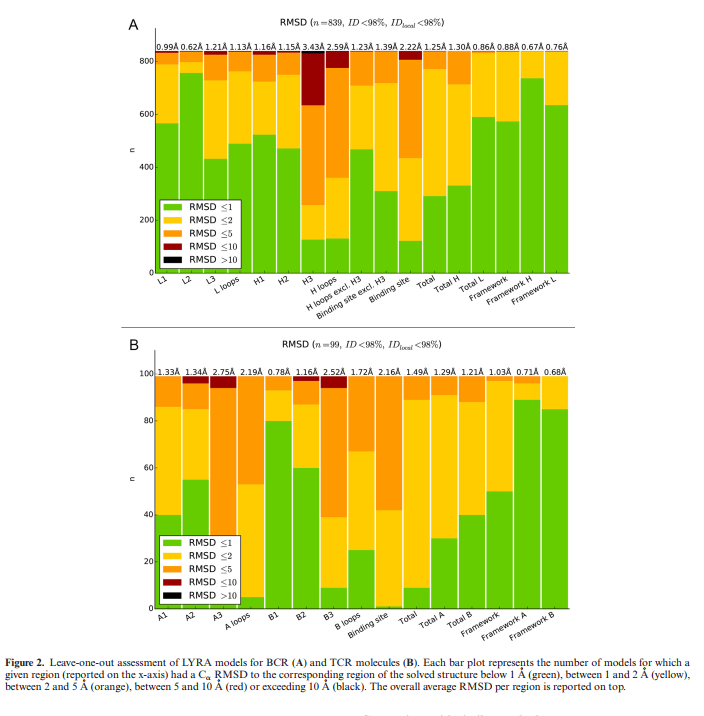
为了测试LYRA的准确性,我们对模板数据库中所有成对的TCR和BCR分子应用了留一法程序。在每一轮中,我们从数据库中删除正在建模的分子以及与该分子本身具有90%或更高序列相似性的任何模板。评估的总体结果如图2的A和B所示。对于TCR和BCR建模,服务器的准确性很高(TCR:1.48Å全局RMSD,2.13Å结合位点RMSD和BCR: 1.29Å全局RMSD,2.24Å结合位点RMSD)。 BCR建模的值与使用最先进的抗体结构预测工具获得的值相同(33)。有关方法准确性的更多详细信息,请参见补充材料(补充图S4和补充表S6和S7)。
4.1 与类似方法的比较
尽管存在其他几种抗体自动建模工具(18,34–39),但据我们所知,LYRA是第一个专门用于TCR建模的公开可用方法。 最近发表的一篇论文评估了商业上和学术上大多数当前可用的抗体建模软件和服务器的准确性(33)。 我们使用LYRA对本次评估中使用的所有抗体进行建模,并采用了留一法操作中相同的冗余度降低,结果如表1所示。我们可以观察到LYRA以完全自动化的方式构建了准确的模型 与手动策划和优化的模型相当。 仅对于兔抗体Ab01(PDB代码:4MA3),由于其重链FR3区的异常插入,LYRA无法产生模型。

4.2 案例分析
为了演示LYRA Web服务器也用于TCR建模的功能,我们展示了4×6B(40)的建模,该模型是最近在PDB数据库中发布的TCR分子,并且与我们数据库中最接近的模板具有86.5%的序列相似性。 对于任何TCR分子,α链的α含量为90.4%(β链)。 LYRA生成的模型叠加到4x6B晶体结构上,如图1面板B所示。它具有目标结构为1.61Å的整体RMSD,框架叠加后的结合位点RMSD为2.75Å。 从图中可以明显看出,该模型具有总体良好的质量,其中大多数不准确性都集中在两条链的CDR3区。 可以在补充表S8中找到在本工作中任何时候都没有使用的独立的TCR分子上的方法准确性的概述。
四、结论
我们介绍了LYRA网络服务器,这是一种用于BCR和TCR结构预测的全自动方法。据我们所知,LYRA是第一个用于TCR的自动化建模工具。鉴于其极其简单的界面,我们认为LYRA将帮助对生物信息学和计算工具知识有限的研究人员轻松建立可靠的淋巴细胞受体模型。 LYRA生成的模型的质量可与其他方法相提并论,足以使它们在大量生物技术,制药和计算任务中有用。如预期的那样(25,26,36),弱点是重链,α和β链的第三个CDR环的建模,这是因为没有合适的模板环,或者因为采用了基于序列的模板选择方案无法选择它。尽管可以在短期内实现一些改进(例如,通过在模板选择过程中包括相邻CDR之间的预测相互作用),但我们相信,由于越来越多的BCR和TCR解决的结构越来越多,这些问题在未来将不再那么引人注目。变得可用,从而增加了可用模板的数量,并允许开发更复杂的模板选择方法。
参考资料
- Klausen MS, Anderson MV, Jespersen MC, Nielsen M, Marcatili P. LYRA, a webserver for lymphocyte receptor structural modeling. Nucleic Acids Research. 2015 Jul 1; 43(Web Server issue):W349-W355. doi:10.1093/nar/gkv535. PMID: 26007650 。网址: https://academic.oup.com/nar/article/43/W1/W349/2467978
