【4.4.3.3】NetMHCIIpan-4.0
主要的组织相容性复合物(MHC)分子在细胞表面表达,向T细胞呈递肽,这使其在T细胞免疫应答的发展中起关键作用。 MHC分子有两个主要变体:I类MHC(MHC-I)和II类MHC(MHC-II)。
- MHC-I主要呈现源自细胞内蛋白质的肽
- 而MHC-II主要呈现源自细胞外蛋白质的肽。
在这两种情况下,MHC和抗原肽之间的结合都是抗原呈递途径中最有选择性的步骤。因此,肽与MHC结合的预测是预测T细胞免疫应答可能的特异性的有力工具。通常,MHC结合预测工具接受结合亲和力或质谱洗脱的配体(mass spectrometry-eluted ligands)的训练。但是,最近的研究表明,两种数据类型的集成如何提高预测性能。受此启发,我们在这里介绍了NetMHCpan-4.1和NetMHCIIpan-4.0,这两个Web服务器被创建来分别预测肽与MHC-I和MHC-II之间的结合。两种方法都利用量身定制的机器学习策略来集成不同的训练数据类型,从而产生最先进的性能并超越竞争对手。这些服务器位于 http://www.cbs.dtu.dk/services/NetMHCpan-4.1/ 和 http://www.cbs.dtu.dk/services/NetMHCIIpan-4.0/ 中。
安装
下载得到netMHCIIpan-4.0.Linux.tar.gz (需要用学术邮箱申请)
tar zxvf netMHCIIpan-4.0.Linux.tar.gz cd netMHCIIpan-4.0
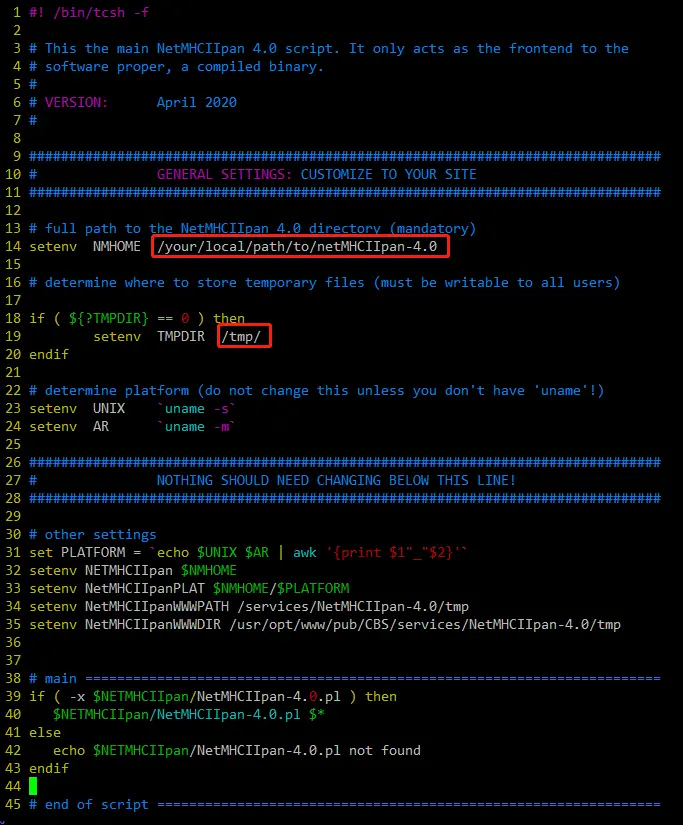
如下图:需要设置2个地方:
3\. In the 'netMHCIIpan-4.0' directory edit the script 'netMHCIIpan':
a. At the top of the file locate the part labelled "GENERAL SETTINGS:
CUSTOMIZE TO YOUR SITE" and set the 'NMHOME' variable to the full
path to the 'netMHCIIpan-4.0' directory on your system;
b. Set TMPDIR to the full path to the tmp directory of you choice (must
be user writable);
一、前言
主要组织相容性复合物(MHC)是脊椎动物细胞免疫系统的基本细胞表面蛋白。 MHC的主要功能是与来源于细胞内或细胞外蛋白质消化的肽(小蛋白质片段)结合,并将其展示到细胞间空间。如果T细胞识别并结合肽-MHC复合物,则可以触发免疫反应,受损的细胞将被裂解。鉴于此,抗原肽与MHC分子的结合代表了细胞免疫的必要步骤,并且了解此事件的规则在人类健康应用中具有巨大而宝贵的潜力。
MHC有两个主要变体:I类MHC(MHC-I)和II类MHC(MHC-II)。 MHC-1在细胞蛋白酶降解后与细胞内蛋白质的肽结合,并作为自身肽组库中抗原变异的控制机制。另一方面,MHC-II结合通过蛋白酶消化细胞外蛋白而产生的肽。这样,两个MHC系统都可以通过将非自身蛋白呈递给T细胞来控制外来生物(1)。考虑到这一事实,已经致力于开发能够精确预测与MHC-1和MHC-II结合的肽的计算方法。
已经使用不同类型的实验数据来训练这些方法。根据此类训练数据的性质,我们可以将肽-MHC结合预测因子分为三大类。
-
第一类对应于接受结合亲和力(BA)数据训练的预测变量(3–6)。此类数据对预测性能施加了实质性限制,因为它仅对肽-MHC结合的单个事件进行建模,而忽略了该过程中涉及的任何其他生物学特征。
-
第二类包括使用从质谱(MS)实验中检索到的数据进行训练的方法,称为洗脱配体(EL)(7-11),或者对BA和EL数据进行整合(5,12-15)。后一种数据类型不仅包含与肽-MHC结合事件有关的信息,而且还包含有关生物抗原呈递途径过程中先前步骤的信息。但是,除了基因工程细胞外,由于多个MHC等位基因变体,细胞MHC表达谱非常不同。同样,用于纯化MS EL管线中肽-MHC复合物的抗体大多是泛特异性或基因座特异性的,从而导致固有的多特异性(或MA等位基因)数据(即,数据包含与多个同源MHC结合基序匹配的肽) )。因此,通常需要先有用户偏见的肽-MHC注释标准,才能解释此类EL MA数据,将其转换为单一等位基因(EL SA或单个肽-MHC注释)并将其用于训练MHC特异性结合预测因子(16)。
-
算法的第三和最后一类试图解决第二种模型的这种局限性,并结合预测算法的训练,具有将EL MA序列注释为单个MHC限制的功能(17,18)。一种这样的方法称为NNAlign_MA(17),在训练过程中,它可以使用称为伪标记的策略将具有模糊关联MHC的EL序列聚集成单个MHC特异性。这不仅可以发现新颖的motif ,而且可以极大地扩展训练集的大小,从而可以全面提高该方法的预测能力。
在这项工作中,我们部署NNAlign_MA来更新NetMHCpan和NetMHCIIpan,从而增强其训练能力并提高其预测性能。为此,我们将NNAlign_MA合并到新模型的核心中,从而使我们可以极大地扩展其训练集。进一步讲,我们对两个模型都进行了完全独立的表位评估,并展示了更新后的方法如何胜过其他当前的最新算法。 铁汉 11:43:02 NetMHCpan和NetMHCIIpan的更新版本在两个关键方面不同于其前身:培训数据和机器学习建模框架。通过累积来自公共领域的MHC BA和EL数据,极大地扩展了培训数据。特别地,EL数据被扩展为包括MA数据。用于训练NetMHCpan-4.1的组合数据集包含13245212个数据点,涵盖250个不同的I类MHC分子,用于训练NetMHCIIpan-4.0的组合数据集由4086230个数据点组成,涵盖了116个不同的MHC II类分子。有关训练集和数据分区的特定详细信息,请参阅补充材料。
机器学习框架已从NNAlign更新为NNAlign_MA,以有效处理这些MA数据。简而言之,NNAlign框架是一个单等位基因框架,允许在模型训练中集成混合数据类型(BA和EL),从而可以在不同数据类型之间利用信息,从而提高了预测能力(12, 13)。 NNAlign_MA扩展了该训练框架,以允许合并EL MA数据。这是通过在模型训练过程中向MA数据迭代注释最佳单等位基因来实现的,从而有效地使MA结合基序反卷积(17)。有关模型超参数和交叉验证训练性能的特定详细信息,请参阅补充材料。
二、网页界面
2.1 提交页面
输入数据
两台服务器都接受两种不同类型的输入。 FASTA和肽。输入数据可以直接粘贴到提交框中,也可以从用户的本地磁盘上载。对于FASTA输入,用户可以指定要包含在预测中的肽段长度(对于I类,长度范围为8至14个氨基酸,默认值为8-11;对于II类,仅允许一个长度(默认值为15)。
同样,对于II类,可以指定是否要使用CONTEXT编码(13)。该上下文由跨越配体的源蛋白质N和C末端部分的氨基酸组成。
提交页面包括所有接受格式的输入数据示例,并提供用于自动上传样品数据的按钮。
MHC选择
接下来,服务器提供一个下拉菜单,以便选择要使用的MHC家族和分子。 NetMHCpan-4.1涵盖超过11000个MHC分子,涵盖人类(HLA-A,HLA-B,HLA-C,HLA-E,HLA-G),小鼠(H-2),牛(BoLA),灵长类(Patr) ,Mamu,Gogo),猪(SLA),马(EQCA)和狗(DLA),以及NetMHCIIpan-4.0涵盖了将近1000人(HLA-DR,HLA-DQ,HLA-DP)和小鼠(H -2)MHC等位基因。对于DQ和DP,用户可以组合覆盖的α和β蛋白链。此外,鉴于两种方法的泛特异性,可以通过上载FASTA格式的全长MHC蛋白序列,对已知序列的任何MHC分子进行预测。
附加配置
两种NetMHCpan方法都基于%Rank分数来告知序列是强MHC结合物(SB)还是弱MHC结合物(WB)。简而言之,%Rank是一种转化,可标准化不同MHC分子之间的预测得分并实现种间MHC结合预测比较。通过将查询序列的预测分数与所讨论的MHC的预测分数分布进行比较,可以计算出查询序列的%Rank,该分布是根据一组随机天然肽估算得出的。鉴于此,%Rank值为1%意味着查询的序列获得的预测得分与从随机天然肽获得的最高1%得分相对应。可以通过指定相应的阈值来修改用于检测SB和WB的%Rank值(默认情况下,将考虑将%Rank <0.5%和%Rank <2%阈值用于检测I类的SB和WB和%Rank <2%和对于II类的SB和WB,%Rank <10%)。此外,如果选择FASTA作为输入格式,则可以选择仅报告低于定义的%Rank阈值的序列,并且对于II类,仅打印与给定结合核心重叠的最强结合肽。
另外,用户可以选择获得输入序列的BA预测分数以及EL可能性,并根据相应的EL预测值(从高到低)对输出进行排序。另外,为了方便用户,可以将输出另存为*。还提供了XLS文件(大多数电子表格软件均可读取)。
2.2 输出页面
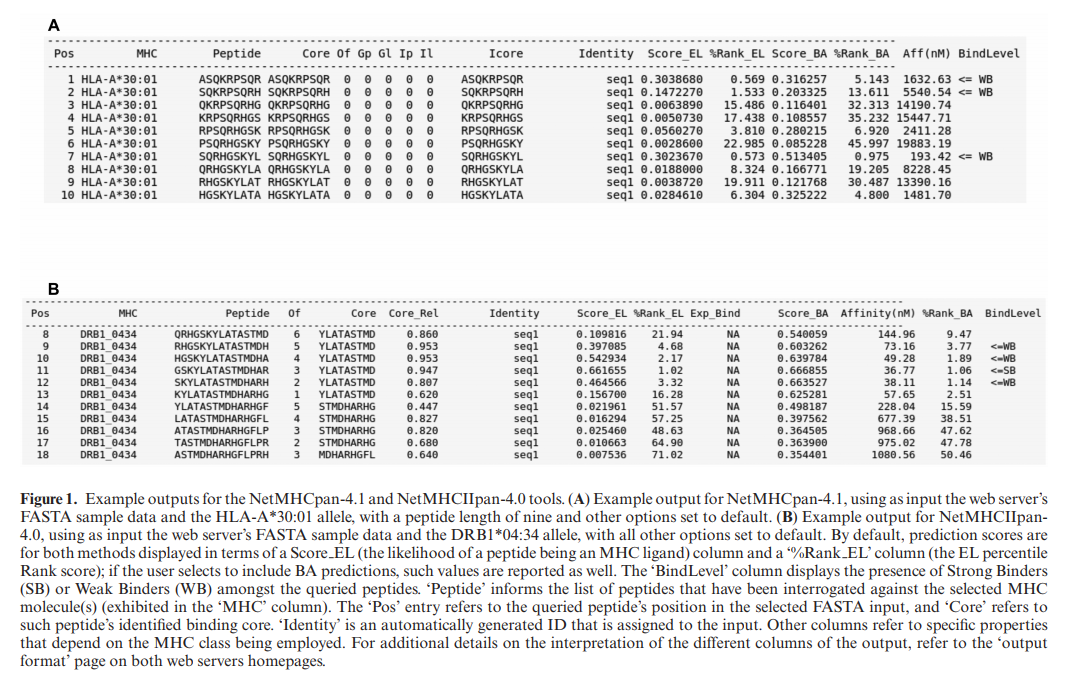
来自两个服务器的输出详细说明了所选MHC分子提供的输入序列的结合预测值,以及指导结果解释的附加信息。 如图1所示,NetMHCpan和NetMHCIIpan输出由几个纯文本列组成,这些列显示有关预测结果的不同信息。
三、评估与范例
作为独立的验证,这些模型以T细胞表位数据集为基准,而I类也为EL SA数据。对于I类MHC,表位数据集取自Jurtz等。 (12)与从IEDB获得的,经MHC多聚体验证的综合表位组合,以及从Reynisson等获得的II类MHC。 (19)。 EL SA数据是从(20)获得的。在所有情况下,都对数据进行过滤以确保与训练数据不重叠(有关数据集的更多详细信息,请参见补充材料)。对于表位数据,根据FRANK(12)估算了预测性能。也就是说,对于每个表位-HLA对,使用洗脱的配体似然预测得分预测了源蛋白的所有重叠肽与HLA的结合,并且报告了FRANK值是预测得分高于HLA的肽的比例。使用此度量,值0对应于完美的预测(在源蛋白内发现的所有肽中,已知表位具有最高的预测结合值),而值0.5对应于随机预测。此外,报告了每个表位的相应AUC,再次将源蛋白中除表位以外的所有重叠肽分配为阴性。有关CD8表位基准的更多详细信息,请参见补充表7。对于EL SA数据集,如“材料和方法”中补充材料的“培训和测试数据”部分所述,添加了阴性诱饵肽。根据AUC,AUC0.1和PPV进行评估。在这里,PPV是根据前N个预测中的阳性肽比例估算得出的,其中N等于配体总数乘以0.95(以考虑潜在的MS污染物)。有关EL SA基准的其他信息,请参阅补充表8。
这些基准的结果如图2所示。在此,将NetMHCpan-4.1与NetMHCpan-4.0(12),MixMHCpred(18,21),MHCFlurry(5)和MHCFlurry_EL(使用EL SA数据训练的MHCFLurry的未发布版本)进行了比较。 ,可在GitHub(22)上找到)。 对于此基准,由于MixMHCpred无法预测包含“ X”(通配符氨基酸符号)的肽,因此将这些肽从基准数据集中删除。 以类似于NetMHCIIpan-3.2(23),MixMHC2pred(11),MHCnuggets(24)和DeepSeqPanII(25)的方式比较了NetMHCIIpan-4.0。
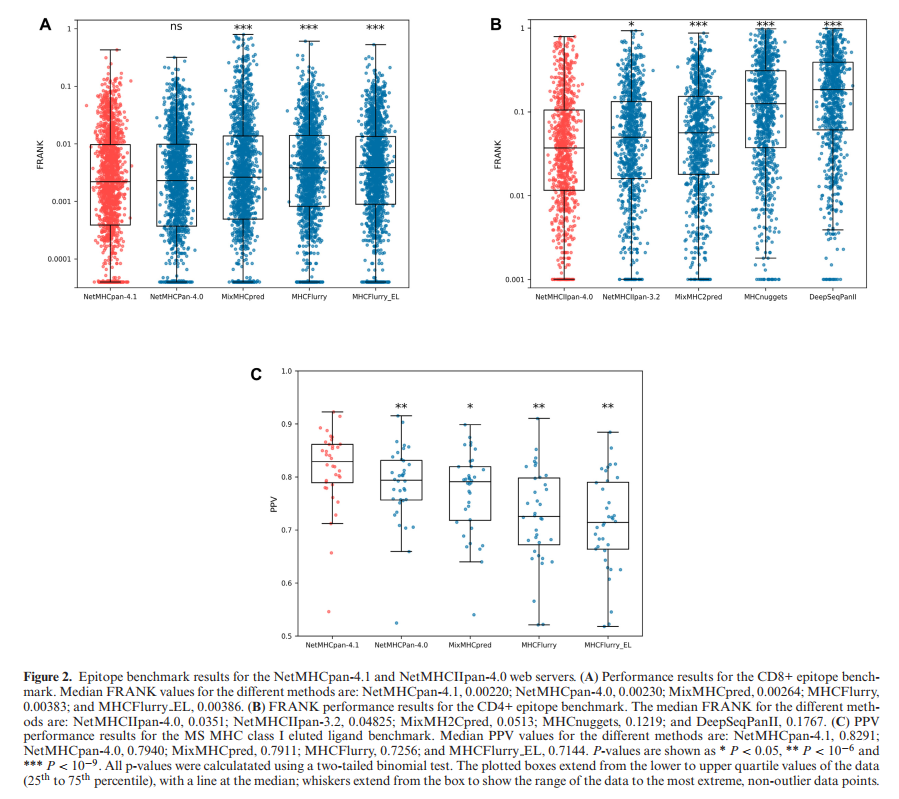
除了在表位基准测试中测试NetMHCpan-4.1和NetMHCpan-4.0之外,所有三个基准测试均证实NetMHCpan-4.1和NetMHCIIpan-4.0的性能明显优于各自基准测试中包含的所有其他方法。 对于I类表位基准,发现NetMHCpan-4.1和NetMHCpan-4.0具有可比的预测性能。 与NetMHCpan-4.0相比,对于NetMHCpan-4.1,在表位和配体基准方面HLA-B和HLA-C分子均得到了持续改进(与用于训练的EL数据集对这些基因座的覆盖率大大提高一致) NetMHCpan-4.1)。 还要注意,与评估洗脱的配体数据的性能时观察到的结果相反(19),但与早期工作(13,19,26)一致,当包含上下文时,观察到NetMHCIIpan-4.0的性能下降 信息(补充图S3)。
四、讨论
在过去的几年中,已经有了大量新颖的MS洗脱的MHC配体数据,从而可以高度丰富地表征MHC呈现的配体组。在这里,我们受益于此数据,并将其与IEDB中提供的大量MHC肽结合数据集相结合,开发了NetMHCpan和NetMHCIIpan工具的更新版本。两种方法都能够预测肽向MHC I类和II类分子呈递抗原(和BA)的可能性。两种工具均使用NNAlign_MA机器学习框架进行了培训,该框架可整合从表达多个MHC等位基因的细胞系获得的MS配体数据集。这些方法相对于其他可用的最新技术的基准测试显示出显着提高的预测能力,可预测MHC配体和T细胞表位。
对于NetMHCpan-4.1和NetMHCIIpan-4.0而言,发现性能提升对于MS鉴定的MHC配体的预测最为明显。这尤其适用于I类,在该类中,发现表位基准上的NetMHCpan-4.1方法与其最新祖先NetMHCpan-4.0的性能相同。存在这种对表位预测性能的有限影响的可能原因,包括当前可用于过去的预测方法和体外实验验证技术的表位数据中的偏差,以及与T细胞表位不共享的MS EL数据中的偏差。未来的工作将解决这些偏见的影响和重要性,并使我们能够将预测MS MHC配体的能力提高到何种程度,也可以预测T细胞表位的能力提高到何种程度。
对工具的基准评估表明,NNAlign_MA机器学习框架具有强大的整体强大功能,可以对训练数据中包含的所有MHC分子进行模体去卷积。但是,结果还指出,以有限的配体数据集(例如HLA-C和HLA-DQ)为特征的MHC分子性能较低。尽管从这两个基因座注释为MHC的配体数量如此之低,部分可以从它们相对较低的蛋白质表达来解释,但其他原因可能包括在纯化MHC分子时用于免疫沉淀(IP)的抗体的HLA-局部特异性不同进行MS实验。未来的工作可能会告诉我们,使用具有改进的HLA-DQ特异性的抗体还是使用具有标记HLA分子的工程化细胞系,如(8)所建议的,可以帮助解决这一问题。
即使对此处提出的预测方法(以及其他最近发布的方法)的改进性能的主要贡献之一是集成了MS衍生的EL数据,但MS数据本身包含固有的偏差,导致例如“可飞行”(flyable)的过分表示(27)和忽略含半胱氨酸的肽(7)。这些偏差对MS中可检测的配体集施加了限制,因此对学习的结合基序也产生了限制。鉴于此,可能需要进一步互补的技术平台来进行MHC肽相互作用的高通量检测,以完成我们对HLA抗原呈递的理解。
NetMHCpan和NetMHCIIpan都具有易于使用的用户界面,允许简单上传查询序列数据,并查询要结合的MHC等位基因。 作为当前唯一公开可用的工具,这两种方法均显示出真正的泛特异性功能,使用户可以预测所有MHC分子,包括以前没有结合数据表征的分子。 工具的输出以简单文本格式提供指导信息,帮助用户选择相关的表位/ MHC-配体候选物。
鉴于已证明的高性能和易用性,我们希望更新的Web服务器将成为指导未来合理表位发现项目的相关工具。
参考资料
- https://academic.oup.com/nar/article/48/W1/W449/5837056 Nucleic Acids Research, Volume 48, Issue W1, 02 July 2020, Pages W449–W454. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data
- https://www.jianshu.com/p/ee707e54065e
