【4.2.1】paratope多样性
抗体分子能够以高亲和力和特异性识别任何抗原。为了深入了解这种功能多样性来源的分子多样性,我们汇编并分析了227个抗体 - 抗原复合物结构的非冗余比对集合。通过计算丙氨酸扫描量化所有残基侧链的结合自由能,允许第一次大规模定量描述抗体互补位(paratopes)。这表明在30个关键位置中少至8个残基足以解释大多数复合物中80%的结合自由能。在这些位置,残基分布不仅与其他表面残基的分布不同,而且还取决于侧链在相互作用中所起的作用,参与结合能的残基主要是芳香族残基,否则是Gly或Ser。为了质疑这些结合特征的一般性,我们通过噬菌体展示使用具有仅两个多样化互补决定区的偏向合成库分离抗体片段,并解析其与其抗原复合的结构。尽管存在这种有限的多样性,但该结构证明所有互补决定区都参与了与抗原的相互作用,并且源自天然抗体库的规则适用于该合成结合物,从而证明了我们的结果的稳健性和普遍性。
一、前言
抗体分子能够识别任何抗原分子的几乎任何部分。 这种独特的特性使抗体成为研究,诊断和治疗中最常用的试剂之一。 随着应用数量的增加,以及抗体在亲和力,特异性,稳定性和溶解性方面的改进需求,需要更详细地提炼抗体 - 抗原识别知识,以增强合理设计。
互补位定义为抗体与其靶抗原相互作用的部分。因此,它仅针对给定的抗原 - 抗体对进行定义,而不是本身实体。例如,已经显示一些抗体与不同的和不相关的抗原相互作用,因此含有两个不同的互补位。然而,在结合模式中存在一些共同特征,其允许互补位部分地独立于抗原定义。抗体可变区(Fv)可以分为高变区和更保守的区段,称为框架区(FRs)。因为大多数多样性位于抗体表面包装在一起并与抗原大多数接触的高变区,这些高变区通常被认为与形成互补位(paratope)的互补决定区(CDR)相同。这确实是正确的,因为通过仅交换它们的高变区可以在抗体分子之间转移结合活性。然而,高变区的许多残基决不会直接与抗原相互作用,只起结构作用,FR中的几个残基也可能与抗原接触。由于水分子在结合复合物的稳定性中的重要性,互补位的定义进一步复杂化。
抗体及其抗原必须在形状和化学性质方面具有互补表面以实现高亲和力。 形状多样性主要不仅归因于环长度和构象的变化,而且还归因于重链可变结构域(VH)和轻链可变结构域(VL)的相对方向的修饰[15], 以及绑定界面的溶剂化。 这种形状互补性涉及许多芳香族残基,它们主要使用范德华力和疏水相互作用将两个表面结合在一起。 通过带电侧链之间的静电相互作用以及桥接氧和/或氮原子的氢键获得特异性和强化。
一些研究已经分析了大量抗体 - 抗原复合物以精确描绘互补位和表位。然而,这些研究中的大多数通过比较存在和不存在结合抗原时抗体残基的可及性来定义相互作用的残基。如果残留物被埋在相互作用的界面内,则将其视为接触残留物并参与相互作用。然而,溶剂可及表面积的变化并不总是与结合的自由能相关。在其他研究中,作者使用距离截止来定义互补位残基。例如,在一系列论文中,Ofran博士的研究小组通过考虑两个残基是否接触来确定200个抗体 - 抗原复合物中的相互作用残基,如果它们彼此相距至少有一对原子。两种方法都具有以大量假阳性位置为代价,给出潜在互补位残基的详尽列表的优点,因为抗体-抗原界面内直接接触的残基不一定参与结合自由能。
在这项工作中,我们专注于对大量抗体 - 抗原复合物中的侧链贡献进行定量和统计分析。我们首先建立了227个非冗余结构的数据库,我们使用计算丙氨酸扫描程序计算了结合界面中每个残基对抗体 - 抗原相互作用自由能的贡献。为了比较结构之间的结果,我们使用先前建立的编号方案对所有抗体进行编号,该方案最小化了对齐结构域的平均结构的平均偏差。这使我们能够定量地定义残基平均贡献,重新审视互补位定义,并解决有关重要联系数量,CDR相对自由能贡献以及侧链使用和角色的问题。该研究表明:
- 互补位主要是不连续的,包含位于30个固定位置的4至13个残基之间,
- 并且含有与在抗体表面的其余部分发现的氨基酸不同的氨基酸子集。
最后,我们解析了抗体 - 抗原复合物的结构,其抗体先前已经从仅具有两个多样化CDR的偏向合成抗体库中分离出来,并且我们证明了这种非天然抗体分子符合我们基于天然抗体的分析中获得的规则。总之,我们的结果揭示了保守的结构限制,其形成抗体互补位的有限多样性,是天然或合成来源,并且应该有助于理解免疫识别的机制和新的和改进的抗体的合理设计。
二、结果
2.1 抗体侧链结合自由能与抗原的对齐数据库
为了本研究的目的,我们关注蛋白质和肽抗原,因为已知半抗原具有不同的结合模式,需要独立分析以得出一般特征规则。我们首先从IMGT数据库(补充表1)中回收了506个抗体 - 抗原复合物结构,去除了错误注释的结构,然后过滤了序列冗余。我们独立地从结构中提取VH和VL结构域以及抗原的相互作用分子。最后,我们使用通用命名方案重命名抗体和抗原链,并使用AHo编号重新编号可变域。基于已知的免疫球蛋白结构域三维结构的空间排列,AHo编号的选择取决于其设计。最终数据库包含227个VH和206个VL非冗余重编码结构及其相应的相互作用抗原分子(补充表2),代表206 Fv和21 VHH。为了评估结合界面中存在的每个抗体侧链对结合自由能的贡献,我们使用FoldX程序进行了计算丙氨酸扫描实验[22]。每个残基侧链对433复合物的抗体 - 抗原结合自由能的贡献在Supplementary File complexes.xlsm中给出。
2.2 很少残基对抗体 - 抗原结合自由能有显着贡献
对于每种结构,我们按其结合自由能的降序对残基进行排序,并计算得到的结合自由能的百分比,作为重链(a),轻链(b)和残基的残基数的函数。完整的Fv域(c)(图1)。具有最高抗体 - 抗原结合自由能的侧链平均分别对VH,VL和Fv的ΔΔG贡献22%,33%和16%。作为一般趋势,自由能贡献随着排名呈指数下降,因此只有前几个残基对结合自由能有显着贡献。例如,对于VH,VL、Fv分别仅有6个(IC95%:2-10),4个(IC95%:1-6)和8个(IC95%:4-13)残基获得了80%的结合自由能。换句话说,80%的结合自由能是由于95%的Fv-抗原复合物中仅4-13个残基。因此,这代表由与抗原在能量上有利接触的残基形成的功能互补位的长度。结果与轻链类无关,λ和κ可变区的接触数相同(补充图1)。这证明抗体互补位仅需要少量CDR的残基以提高高亲和力和特异性。
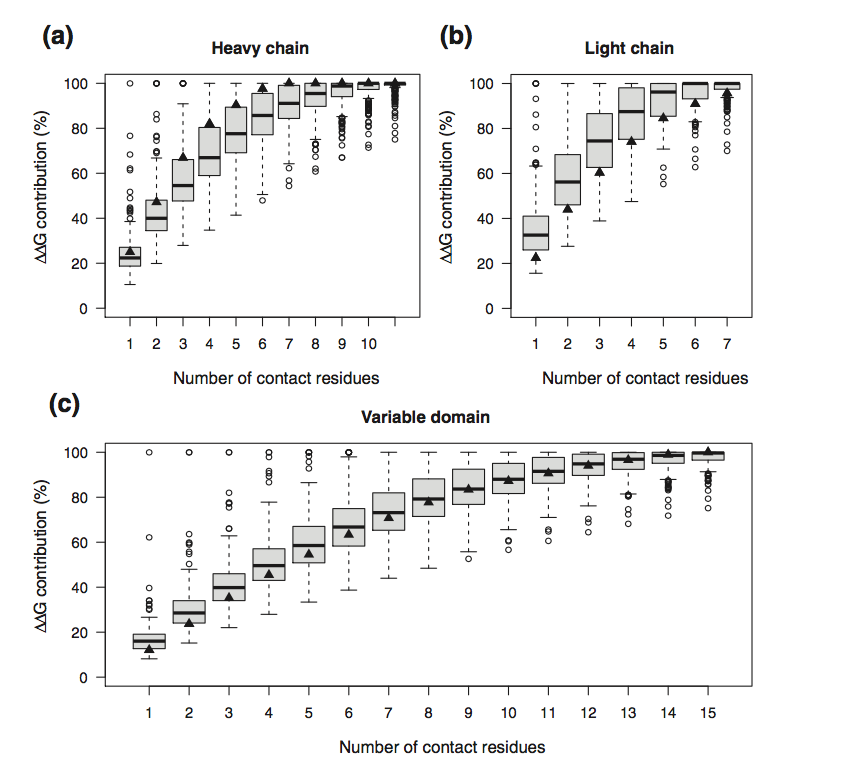
图1 对于VH(a),VL(b)和Fv(c)结构域,对于给定数量的接触残基获得的结合自由能的百分比,分配参与结合自由能的数量。 黑色填充三角形是GkF5复合物的值。 Boxplot胡须扩展到最极端的数据点,不超过四分位数范围的1.5倍。
2.3 每个位置绑定自由能
在先前的结果中,所考虑的残基是不同的并且针对每种抗体 - 抗原复合物进行了最佳选择。为了确定一些常见残基是否参与相互作用,我们计算了一系列复合物中每个位置的平均结合自由能。这可能归功于AHo编号方案,该方案确保同等数量的残基也是结构等价物。详细结果显示在Supplementary File complexes.xlsm中。对于重链和轻链,图2中显示了每个位置的平均结合自由能的图。大多数相互作用位点位于CDR区(Kabat的定义),但一些常用位置如H2,H29-32,H44,H54,H108,H139,L2,L44,L54和L57位于FRs中(平均ΔΔGN0.1千卡/摩尔)。在CDR内,抗体 - 抗原结合自由能的不均匀分布限于几个特定位置(hotspots)。实际上,分别只有2,4,1,2和2个位置对H1,H2,L1,L2和L3的总CDR结合自由能的贡献超过60%。我们注意到,仅在H3中,结合自由能更均匀地分布。这是由于该CDR的长度变化很大,可以平滑每个位置的能量。如果已经针对任何给定长度研究了H3环中的能量分布,则对于其他CDR(补充文件complex.xlsm),分布将变得更加异质。然而,由于H3环结构的多样性非常高,除了环的N-和C-末端部分外,等数残基的结构等同性是非常值得怀疑的。根据它们的平均ΔΔG对位置进行排序,并且将总ΔΔG的百分比作为这些固定残基的数量的函数示于图3。这表明,例如,总ΔΔG的80%位于30中。对于一半以上的结构,CDR中的27个位点和3个FR中的位置仅占CDR总位置的一小部分(根据Kabat的定义,不到CDR位置的一半)。此外,这些位置中的一些并不总是被占用,并且对于大多数抗体而言,它们的数量可以容易地减少。结果如图1和图2所示。对于主要包含κ轻链的整个数据库获得2和3。如果独立分析,最佳位置略微依赖于轻链类,λ轻链具有比κ轻链更强的倾向以使用残基L109结合抗原,κ轻链具有经常更长的L1环与残基L32-L36接触(补充图2)。
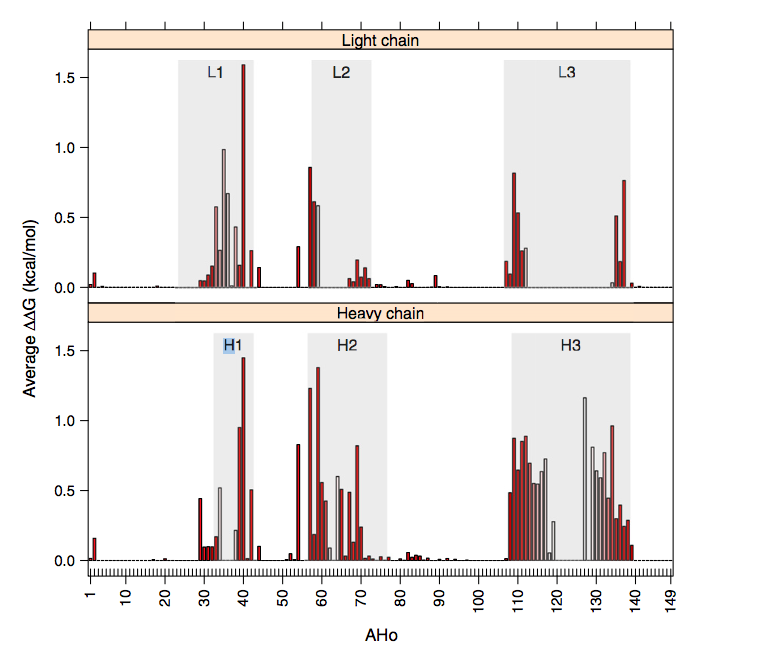
图2。 Average binding freeenergy.Bars表示VL(顶部)和VH(底部)结构域的平均抗体 - 抗原结合的自由基能。 条形颜色表示在比对抗体的数据库中该位置的占据频率,从0%(白色)到100%(更深的红色)。
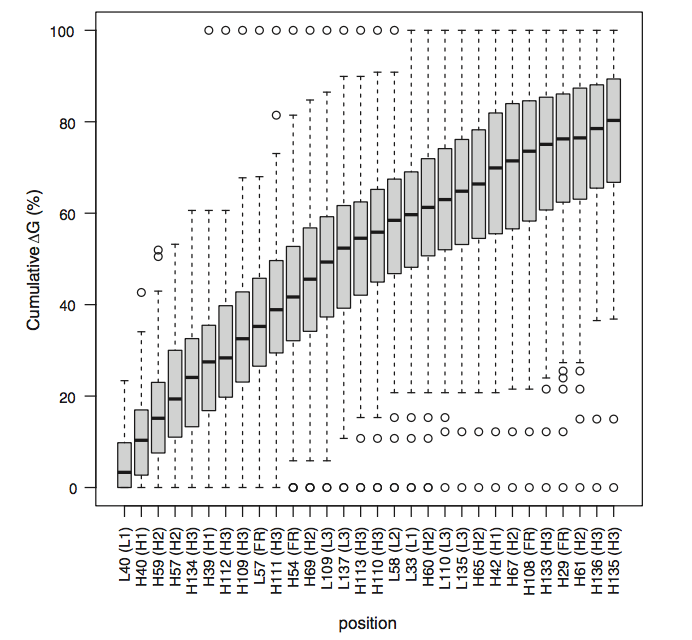
如图3所示。 累积结合自由能量。分配自由的能量到30个贡献位置。能量从左边累积。 仅显示超过40%的结构占据的位置。 Boxplot胡须扩展到最极端的数据点,不超过四分位数范围的1.5倍。
2.4 CDR对抗原结合的贡献
计算206个Fv-抗原复合物的每个CDR的结合自由能贡献(图4a)。 H2和H3是对抗体 - 抗原相互作用贡献最大的两个CDR,它们中的每一个在一半复合物中占总结合自由能的22%。三个CDR H1,L1和L3几乎同等地贡献相互作用的自由能,中值分别为10%,9%和12%。最后,L2在超过一半的复合物中不与抗原相互作用。 此外,在每个复合物中估计参与抗原结合的CDR的数量(图4b)。当CDR的至少一个残基显着参与相互作用时(ΔΔGN0.8kcal/ mol),认为CDR与抗原相互作用。使用的CDR数量的分布显示,在97%的案例中,至少三个CDR含有一个或多个这样的重要残基。值得注意的是,在该分析中使用的206种复合物都是具有重链和轻链的标准抗体,丢弃了VHH样抗体,其中仅由三个CDR限制的相互作用模式非常不同(补充图3)。
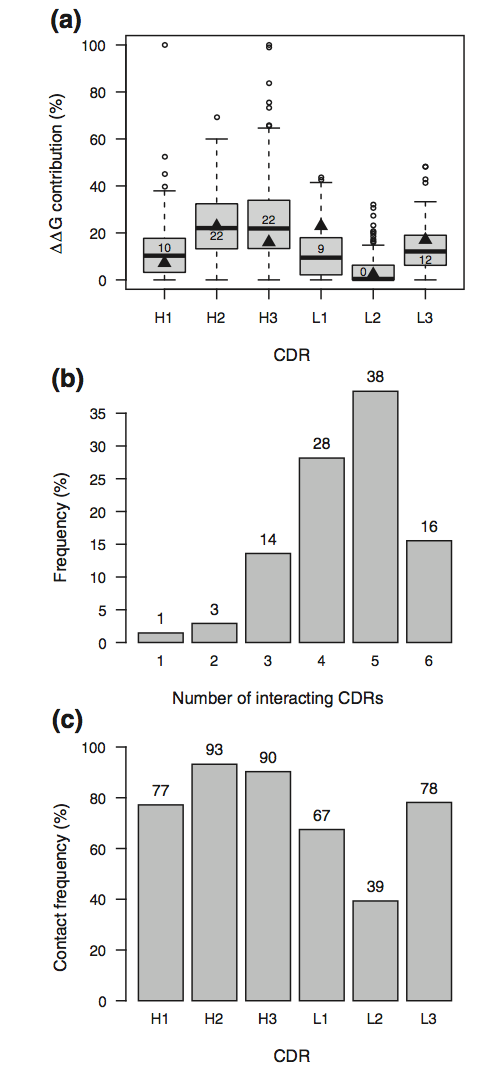
图4. CDR贡献和用法。 (a)CDR结合自由能贡献的分布。 方框中的值是分布的中位数。 黑色填充三角形是GkF5复合物的值。 Boxplot胡须扩展到最极端的数据点,不超过四分位数范围的1.5倍。 (b)具有给定数量的相互作用CDR的结构的百分比。 (c)指示的CDR有助于结合自由能的结构的百分比。
与每个CDR的结合自由能贡献相关(图4a),H2和H3含有至少一个对结合能显着贡献的残基(ΔΔGN0.8 kcal / mol)在93%和90%的 分别是抗体 - 抗原复合物,而只有39%的结构在L2和抗原之间显示出显着的相互作用(图4c)。 最后,H1,L1和L3分别含有具有显着游离结合能的残基,分别占77%,67%和78%。
2.5 侧链使用和角色
一些研究表明,特定氨基酸侧链在蛋白质复合物形成过程中起着不同的作用,包括抗体。鉴定了抗原结合中涉及的抗体位置及其平均结合自由能,我们重点详细分析了侧链侧链相互作用过程中每种侧链类型所贡献的能量。 Paratope分析限于15个最有贡献的CDR位置,其代表约50%的结合能(图3)。作为参考,我们使用了62个从未与抗原相互作用的表面残基(图2中的平均ΔG= 0和溶剂可及性N 19%)。对于这两类表面残基,侧链使用频率非常不同,说明抗体hotspot处的高度特异性残基使用(图5)。在这两组中最常见的7个残留物中,占所用残留物的75%,只有两个残基是共同的,G和S酪氨酸是热点位置最常见的残基,频率为24%,仅存在于在非互补位表面位置的频率为2.7%。这种总体趋势在互补位的大多数位置被发现,但是H39,L40和L137显示出稍微不同的分布,前者在Tyr中强烈富集,后者在Pro和Leu中强烈富集(补充图4)。

图5.在paratope和非paratope位置的残基使用。 条的高度是在互补位的15个最大贡献位置(左轴)或在62个溶剂可及的和非相互作用的残基(右轴)处的给定残基的频率。 深灰色条是所考虑的残基的频率,其对结合的自由能贡献至少0.8kcal / mol。
我们利用数据库的定量性质来计算每个残基对于结合自由能的正贡献的频率(图5中的黑条)。 残基的排名明显不同,可以确定两类:
- 与抗原频繁相互作用的残基,基本上是芳香族化合物,
- 以及在互补位中经常发现但在结合自由能中没有任何作用的中性残基,如 G和S.
最后,对于在互补位上比在其他表面位置更常见的残基,我们注意到它们的侧链经常与抗原相互作用。 例如,Y和W分别富集9和450倍,分别与抗原的66%和74%相互作用,其他3倍富集的残基也参与复合物的稳定性,频率为 至少25%(N:25%,H:37%,D:31%,F:32%)。 另一方面,在互补位上比在蛋白质表面上低3倍的残基Q,K,P和T也不太可能与抗原产生有效的相互作用(Q:22%,K:22%) ,T:19%),脯氨酸除外(51%的生产性相互作用)。 在热点位置脯氨酸残基的这种异常降低的频率可能是由于该氨基酸在不需要其形成互补位时对CDR构象施加的结构限制。
2.6 特征规则的普遍性,结构案例研究
存在于当前蛋白质数据库(PDB)中并且上文分析的大多数抗体已经从免疫动物和由人类供体制备的抗体噬菌体文库中获得。在这两种情况下,互补位的多样性是自然的,并且起源于V基因多样性,V(D)J重组和体细胞超突变事件。我们证明这些抗体使用非常有限数量的残基与它们的抗原相互作用,无论是在位置还是在自然界中。这种受限制的多样性可能源于免疫系统或抗体互补位本身的结构所施加的限制。为了解决这个问题,我们通过对Gankyrin抗原进行淘选,从合成的强偏向抗体文库中分离出单链Fv(scFv)。该scFv以中等亲和力结合其抗原,使用Biacore估计其为100-200nM,通过竞争ELISA测定为450nM 。然而,该复合物足够稳定,可通过凝胶过滤色谱法与游离抗原和scFv分离。在使用的文库中,仅在两个CDR3中引入了多样性,其他四个CDR固定于抗β-半乳糖苷酶抗体的那些。另外,虽然引入的H3序列接近人类序列,但引入的L3是不自然的,因为其N-末端部分不对应于任何天然种系。然后,我们解析并分析了Gan-kyrin-scFv F5(GkF5)复合物,以确定文库设计是否可能偏向于互补位置和化学成分。
GkF5的晶体结构以2.5Å的分辨率求解(表1)。 Gankyrin由七个锚蛋白重复序列ANK1-ANK7组成。 它的符合性类似于先前解决的Gankyrin结构。 结合和未复合的Gankyrin结构的Cα原子可以用0.95的RMSD叠加,由于scFv结合而没有显示出大的构象重排。 scFv F5采用富含β-折叠的经典抗体折叠,但由于其灵活性,在电子密度图中未见VH和VL链之间富含甘氨酸的连接区域。 抗体F5与ANK4-ANK6重复的α-螺旋相互作用,它们是Gankyrin凸面的一部分(图6a)。

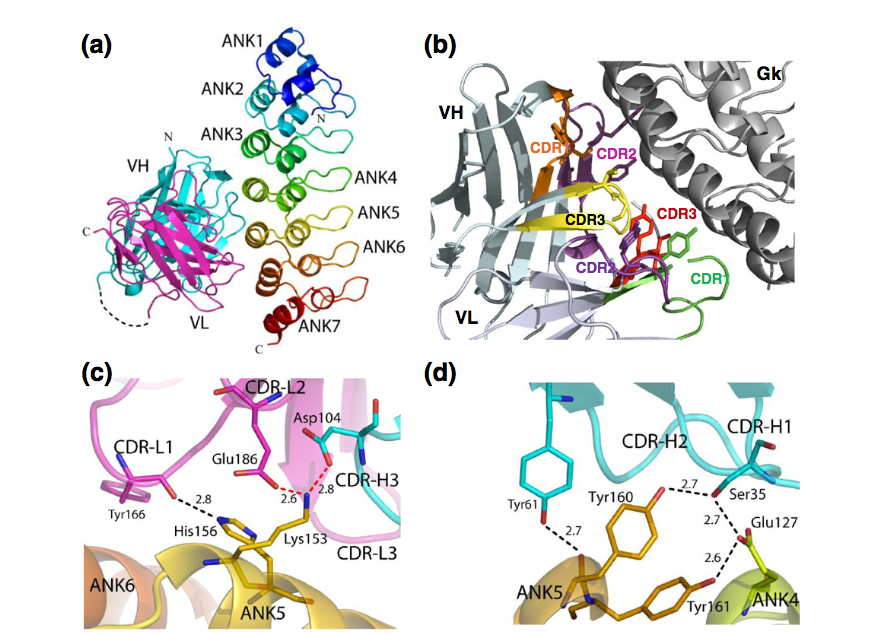
图6。 GkF5结构和分析。(a)GkF5复合物的整体视图。重复Gankyrin的每个ankyr由ANK1-ANK7和不同颜色(从蓝色到红色)表示。 抗体F5的VH和VL结构域分别为青色和品红色。 虚线表示抗体的非结构化连接区域。 (b)GkF5互补位。 表示通过计算丙氨酸扫描鉴定的12个相互作用的残基侧链(表2)。 六个CDR的颜色为橙色(H1),粉红色(H2),黄色(H3),绿色(L1),紫色(L2)和红色(L3)。 scFv F5和Gankyrin之间的离子和氢键表示在VL(c)和VH(d)结构域附近(表2)。 VL,VH,ANK4,ANK5和ANK6重复序列分别为品红色,青色,黄色,橙色和棕色。 氧和氮原子分别为红色和蓝色。 红色和黑色虚线分别表示离子键和氢键。
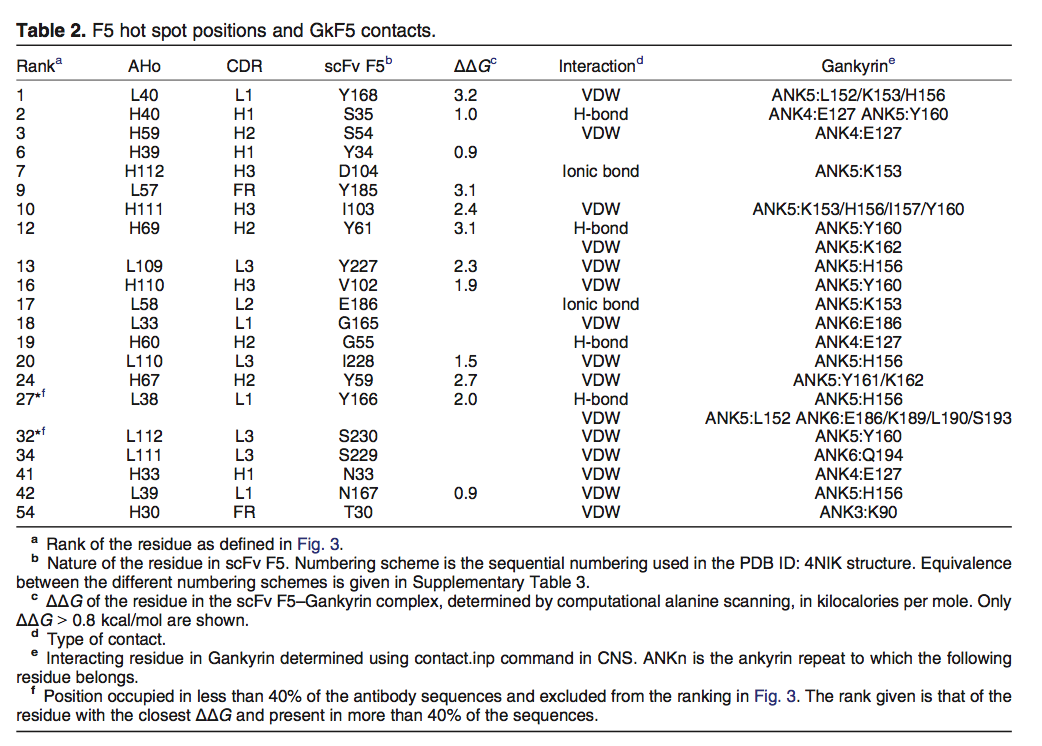
抗体互补位不限于两个多样化的CDR3,而是包括与抗原接触的其他CDR(图6a)。计算丙氨酸扫描分析进一步指出位于5个CDR和1个FR中的12个抗体残基与复合物的稳定性有关(图6b)。这些残基中的11个位于上述从抗体 - 抗原数据库确定的30个最佳热点位置内(表2和图3),并且它们的结合自由能曲线与数据库中获得的平均值很好地拟合(图3中的实心三角形) 1)。遗漏的残基是N167(L39),ΔΔG为0.9kcal / mol。由于其在κ轻链中的低使用,该位置在数据库中具有低平均能量。然而,如果我们仅考虑具有与scFv F5相同的λ类的轻链的Fv结构域,则L39排名第18,平均ΔΔG为0.6kcal / mol(补充图2)。此外,对GkF5复合物的CDR贡献与从数据库获得的平均值非常相似(图4a中的实心三角形)。两个多样化CDR(H3和L3)的结合自由能加起来达到总ΔΔG的32%,而三个相互作用但固定的CDR(H1,H2和L1)和FR对总ΔΔG贡献了68%。由于两个CDR3和scFv的非多样化部分(其他CDR和FR)与抗原形成4和8个接触,因此在接触数中也发现了互补位的固定部分在结合自由能中的更高贡献。最后,83%的总结合自由能是由于天然抗体中仅有8个残基(图1c),其中6个是Tyr。
为了使用不同的方法确认这一结果,我们使用晶体学和核磁共振系统软件套件确定抗体接触,该软件套件使用几何规则来识别相互作用(表2)。 使用计算丙氨酸扫描鉴定的十二个残基中的十个也存在于该分析中,两个遗漏的残基是Y34和Y185。 此外,在19个已确定的残基中,有14个位于图3所示的30个最有贡献位置的列表中。总而言之,这表明使用两种方法,尽管其合成和偏差来源,scFv F5互补位符合一般 我们的数据库分析定义的规则。
2.7 特征规则的普遍性,重要的互补残留的预测
我们接下来测试了我们的数据库是否可用于在没有结构和实验数据的情况下预测重要的互补位残基。作为参考,我们使用了Muller等人生成关于人源化抗VEGF Fab的实验数据。在该文章中,作者将68个CDR残基逐个突变为丙氨酸,并测量所得突变体Fab的相对亲和力。我们将CDR残基分为三类:影响亲和力超过150倍因子的8个主要贡献者,11个残基通过更温和的因子降低亲和力(5至66倍),49中性或弱贡献者。这些残基分别以深绿色,浅绿色和白色背景显示在图7中的Muller1998中。如果我们将这些残基的位置与我们研究中确定为主要热点的位置进行比较(图2) ),穆勒的8个主要贡献者中有7个的平均结合自由能高于0.8千卡/摩尔。由于只有12个热点位置具有如此高的平均结合自由能,这意味着使用我们的数据和针对这12个残基的靶向突变将鉴定出8个Fab主要互补位残基中的7个(88%)。如果我们将结合自由能截止值降低到0.4 kcal / mol,我们的数据库确定了Fab互补位中的27个潜在相互作用位点(图7中的红色和粉红色背景)。在这27个站点中,Muller的数据中还确定了15个位置(中等或主要贡献者)。在这种情况下,只有27个位置的实验性丙氨酸扫描将鉴定出19个互补位残基中的15个(79%)。使用第二抗体进一步证实了鉴定位置和实验数据之间的良好相关性,其中还使用鸟枪扫描诱变分析了整个CDR。同样,在平均结合自由能高于0.8千卡/摩尔的12个位置中,除了两个之外的所有位置在突变为丙氨酸时具有强烈的去除效应(Fwt /ala > 20)(补充图5)。具有高能量且不影响结合的两个位置都在轻链(L40和L137)中,其轻微地参与该特定抗体中的相互作用。此外,在我们的数据库(H33和H135)中,影响结合超过50倍因子的12个位置中只有2个具有低于0.4kcal / mol的平均结合能。
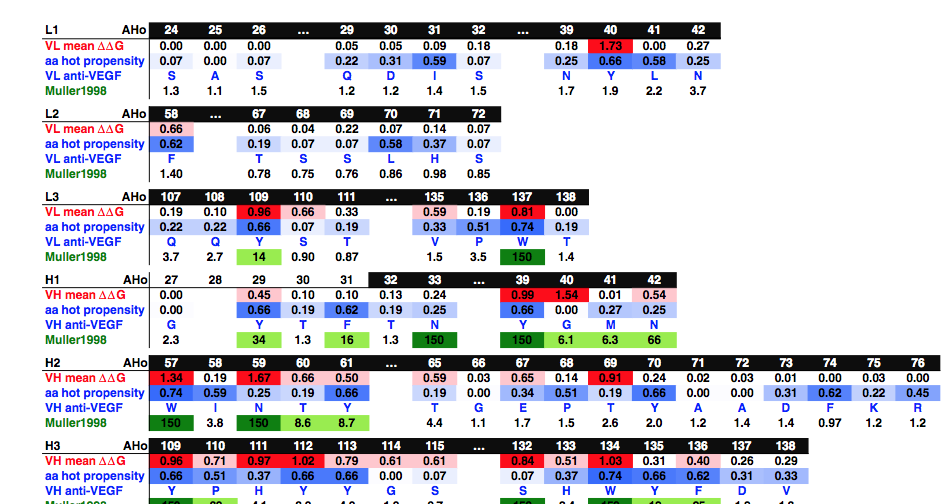
图7.抗VEGF互补位的分析。 使用作为补充材料提供的电子表格“complexes.xlsm”生成数据。 显示了对应于六个CDR并在Muller的文章中诱变的六个区域。 第一行:AHo编号。 黑色背景表示Kabat的CDR。 VH平均值ΔΔG:平均结合自由能(图2)。 ΔΔGN0.8和0.4kcal / mol的位置分别为红色和粉红色。 热倾向:对于所考虑的氨基酸,这是该侧链在15个最强的热点位置处产生能量上有利的键的频率(图5)。 VH / VL抗VEGF:抗VEGF Fab-12的序列。 Muller1998:在Muller出版物中当位置突变为丙氨酸时,结合亲和力的变化。 主要的互补位残留物(亲和力降低150倍)为深绿色,中度贡献者(5至66倍)为浅绿色。
这表明我们编译的游离结合能数据库可用于确定抗体互补位中的重要残基,减少通过定点诱变进行实验测试的残基数量,并增加快速鉴定关键结合残基的机会。
三、讨论
本研究的目的是在分子水平上定量描述抗体分子与其同源抗原结合的模式,确定互补位在位置和性质上的关键残基, 并推导出有助于抗体工程和图书馆设计的一般规则。
在抗体 - 抗原复合物的比对的非冗余结构数据库上的大规模丙氨酸扫描分析使得能够首次定量描述所有抗体残基在结合的自由能中的作用。这些数据提供了对互补位的定量描述,从中可以推导出特征规则。
- 80%的结合能仅由少数显着残基引起,通常为8(4至13之间)。
- 这些残基定位于25-30个固定热点位置,这些位置在超过一半的复合物中占75-80%的能量。
- 3至6个CDR参与复合物形成。
- 热点位置由有限数量的侧链(Y,G,S,W,D和N在61%的情况下)占据,
- 其中基本上芳香族残基有助于结合自由能。
这些规则不仅适用于构成本分析中使用的大多数结构的天然抗体,而且适用于选自具有集中和受限多样性的高度偏向的文库的合成抗体,因此,这些代表了大多数抗体中存在的限制 - 抗原复合物。
由于第一个解析的抗体 - 抗原结构,已知只有一小部分接触残基支配关联反应的能量学。已经使用实验性丙氨酸扫描诱变研究了几种抗体 - 抗原复合物并且精确定量了这些效应。通过使用不同的模型,已经证明相互作用涉及5到10个主要残基贡献者。即使对于抗原具有50pM的高亲和力的抗体,只有4-5个轻链残基对结合能有显着贡献,与我们的数据库中获得的值非常一致(图.1b)。然而,一些已知的抗体偏离该一般规则作为抗独特型抗体E5.2,其中大多数接触残基在结合自由能中起重要作用。这种互补作用的不连续性质可能发挥作用,因为它使抗体对抗原突变更具耐受性,特别是因为新的水分子可以补偿接触的丧失,如在HEL D18A突变体的情况下所证明的那样,与D1.3抗体的相互作用。然而,我们研究的新颖性表明,对于PDB中存在的几乎所有抗体分子存在共同的结合模式。换句话说,不仅少数残基有助于结合自由能,而且这些残基主要位于约25个保守位置。这个有限数量的互补位残基可以解释为什么D1.3与溶菌酶结合,而抗独特型抗体D5.2在17个接触中显示出与13种常见热点残基结合的相似模式。同样,在一些特殊情况下,抗体可能以非常规方式调整其结合,如一些高度突变的交叉中和抗人类免疫缺陷病毒抗体所证明的
一些研究已经分析了抗体分子互补位中侧链的分布,并得出结论,对某些芳香族残基有明显的偏向。然而,在这些分析中,作者使用了跨越大部分CDR残基的互补位的定义。例如,Collis等人分析了它们之前定义为接触残基的位置的氨基酸分布。虽然比传统的CDR短,但这些区域仍然很大并且含有超过50个残基。在我们的情况下,我们将分析限制在大多数分析结构中与抗原形成能量上有利的接触的位置,因为我们仅考虑具有最高平均自由结合能的15个位置(ΔΔGN0.87kcal / mol)。此外,我们使用抗体分子的表面残基作为参考,而Collis使用来自CATH数据库的环区域。尽管存在这些差异,我们仍然认为Tyr和Trp在抗体的互补位中有很强的丰富性。然而,在我们的情况下,这种富集更大,可能是因为这些残基在结合的自由能中的突出作用。还有一些微妙的不一致可能是由于分析的不同残基集。我们注意到富含两种其他芳香族残基(F和H),Asp和Asn,而Collis没有看到这些氨基酸在互补位中有任何显着的富集。对于Tyr和Trp,这种差异可归因于我们分析中对这些侧链的自由结合能的重要贡献。
这里介绍的互补位的不连续性质与传统的CDR定义不同。实际上,大多数主要贡献残基是Kabat高变区的一部分,但事实并非如此,因为这些区域中只有25%贡献超过70%的结合自由能。然而,许多非相互作用残基在避免空间冲突和形成互补位方面起着重要的结构作用。一些研究已经证明了这些残基在抗原结合亲和力中的重要性。但是,由于两个主要原因,它们没有出现在我们的分析中。首先,使用的FoldX脚本将分析限制于抗体和抗原之间界面中存在的残基。其次,这些残基的重要性在抗体分子之间变化很大,我们在227个结构的整个数据库上平均了能量。这些区域在图2中清楚可见,例如,框架位置H82-H84,但它们的贡献仅限于少数抗体。尽管如此,我们的研究表明,相互作用的残基保持广泛保守,只要使用适当数量的环。这里确定的重要位置可能有助于设计几个实验,如抗体 - 抗原建模和对接,亲和力成熟,互补位鉴定,CDR和SDR移植和文库设计。限制已识别位置的多样性并使用针对最重要侧链的遗传密码,应该允许构建更小但更有效的抗体文库,如Sidhu博士的一系列论文中所证明的那样。
作为直接应用,我们显示我们的数据可用于快速查明抗体互补位中的重要残基。然后可以通过实验确认,但只有很少的突变。例如,如图7中所示,在抗-VEGF Fab的情况下,可以仅使用12个突变鉴定了8个最重要的互补位残基中的7个,并且将通过以下方式获得对该互补位的几乎完整描述:仅检测27个位置而不是覆盖6个CDR所需的68个突变(补充图6)。随后可以使用该信息来使用不同的技术或假设来进化抗体以获得更好的亲和力。例如,可以首先使用诱变和选择将所鉴定的位置靶向用于优化,或者相反,可以保持这些位置不变并且仅诱变周围的残基。电子表格可作为补充材料(complexes.xlsm),以帮助读者将自己的抗体序列与我们的数据对齐。
此处提供的许多结果取决于所使用的编号方案。不同方案的相对好处已经在别处讨论,我们决定使用AHo方案有两个主要原因。首先,CDR1的长度变化由两个gap调节,由保守的疏水残基31隔开。其次,插入和缺失对称地围绕关键位置(在补充文件复合体中标记为黄色.xlsm),而在其他编号方案中,插入正在单向增长。这些修饰导致环更好地对齐,并确保结构上等同的残基不仅在FR中而且在CDR中相同编号。但是,长循环提供了更多潜在的交互位点。这在图2中由白色条表示,白色条代表仅存在于长CDR中的热点。这些位置位于环的顶部,在L1和H3 CDR中更常见。然而,存在于最长环中的这些接触通常会增加但不能替代短环中存在的其他接触,导致所考虑的CDR与抗原的更强相互作用。由于H3环结构的非常高的多样性,VH CDR3的情况可能有些不同,但是,某些位置似乎优先用于某些环长度(Supplementary File complexes.xlsm)。
选择粘合剂和库设计的趋势是扩展自然多样性以减轻先天的约束限制。这是有利于合成和半合成抗体文库,而不是天然抗体文库的主要论据之一,以获得针对自身蛋白的抗体。然而,没有实验证据表明这样的合成文库确实比天然组合更能产生这种结合物,并且已经从人类供体的文库中成功选择了几种抗自身抗体。我们通过从合成且强烈偏向的文库中选择scFv并解决复合物的结构,直接测试了逃避天然抗体结合模式的可能性。与通常的预期相反,大多数有约束力的自由能(68%)是由于非多元化的立场。抗体几乎完全满足上面定义的规则,因为大多数相互作用残基位于本研究中鉴定的热点位置,并且CDR贡献类似于从数据库获得的平均值。在最近的一项研究中,Persson等人发现尽管与不同的抗原相互作用,但分离出一系列共享相同H3序列的10个克隆。在我们的案例中,他们表明,在80%的这些克隆中,几个保守的不变H3侧链与抗原相互作用。他们还分离出两种抗体,其中H3环不参与与抗原的相互作用,与我们数据库中10%此类抗体的频率非常一致(图4c)。他们研究的主要结论是可以设计抗体文库以对L3环提供主导作用。虽然这仍然是正确的,但是单独使用,这种抗体存在于自然界中实际上,如图4a所示,在一些抗体 - 抗原复合物中,L3环有助于超过40%的结合能。这再次表明所描述的规则归因于抗体和抗原结构限制,并且即使具有高度偏向和非天然的文库也不容易逃脱。
总之,我们的结果证明了自然界引入的分析方法,即引入抗体互补位的多样性。 由于免疫系统不能对六个CDR(约2060-1078)中随机序列所代表的大量潜在多样性进行采样,因此可能需要将多样性限制在关键位置并使用一组有限的侧链。 然而,我们发现在合成抗体库的情况下也是如此。 我们的分析增加了对抗体 - 抗原相互作用的理解,并可能有助于未来设计具有优化功能多样性的改良抗体和文库。
四、材料和方法
4.1 产生一组抗体 - 抗原复合物
来自PDB的抗体/抗原复合物的结构从IMGT / 3Dstructure-DB数据库(IMGT®,国际ImMunoGeneTics信息系统)中检索。从数据库中检索了总共506个免疫球蛋白抗体或抗体片段的PDB文件(与蛋白质或肽配体复合)(2011年5月的数据)。我们从这些文件中提取了484个VH,404个VL-κ和59个VL-λ序列。 PDB文件数量和提取的抗体序列之间的差异是由于仅包含Fc结构域的文件(缺少可变链),我们从该组中删除了这些文件。使用程序cd-hit 对VH序列进行聚类,使用默认参数和92%的同一性截断值,并在每个簇中保留具有最佳分辨率的序列,得到238 VH,183 VL-κ和30 VL-λ。使用AHo编号方案重新编号所有文件,使用已发表的存在的一组已对齐的可变域、clustalw软件、in-house python脚本,最小化免疫球蛋白结构域的对齐三维结构中残基的平均偏差。 。丢弃与AHo编号不相容的非常规序列,产生234个VH,183个VL-κ和30个VL-λ序列。
当晶体不对称单元中存在多个分子时,仅保留可变结构域的单个拷贝,并分别对于VH,VL-λ和VL-κ重命名为“H”,“L”或“K”(在 -house python脚本)。 使用来自CCP4套件的NCONT程序将配体相互作用链定义为至少与抗体CDR残基接触一次。 丢弃没有任何与可变结构域相互作用的配体链的结构。 对这些特定文件的检查显示,配体在IMGT中被错误注释为半抗原(因此被NCONT忽略)或者相互作用是通过Fc结构域。 最终的抗体 - 抗原数据库由227个VH,175个VL-κ和31个VL-λ组成,包括本研究中解决的结构。
4.2 使用FoldX绑定自由能计算
在改变结合自由能计算之前,使用FoldX 3.0beta6的修复功能优化所有复合物。 在对复合物进行优化后,我们使用FoldX程序的Complex_alascan命令计算每个抗体侧链的自由能,从而有助于抗体 - 抗原相互作用,其中给定抗体的每个位置(甘氨酸和丙氨酸除外) - 将抗原界面截短为丙氨酸,并优化相邻侧链的位置。 结果提供了“突变体”和“野生型”结构之间的结合自由能ΔΔG(以千卡/摩尔计)的差异,对应于界面中存在的每个抗体侧链的结合自由能并且有助于抗体 - 抗原相互作用(分子间键)。
4.3 数据收集,结构确定和改进
如前所述表达并纯化蛋白质。通过混合0.2μl蛋白质溶液和0.2μl含有0.1M Hepes(pH7.0)和8%聚乙二醇8000的储库溶液,在17℃下通过蒸气扩散获得Gankyrin和scFv F5的复合物的晶体。将晶体安装在纤维环中,并在用储液溶液加15%乙二醇冷冻保护后在液氮中快速冷却。来自冷冻单晶的数据收集在欧洲同步辐射装置(法国格勒诺布尔)的光束线ID29上以100K进行。该晶体属于三角形空间群P3221,每个不对称单元具有一个Gankyrin-scFv F5复合物。使用HKL2000 对数据进行整合和缩放(参见表1中的统计数据)。使用程序AMoRe通过分子置换解析结构[56]。 Gankyrin和中和抗体F10 [PDB ID:1UOH和PDB ID:3FKU(链X)]的结构分别用作起始模型。细化涉及手动构建和细化计算的迭代循环。程序phenix.refine 和Coot用于整个结构确定和细化。 scFv F5的接头的几个末端残基和16个残基(119-134)未被建模,因为相应区域中的电子密度图差。使用各向异性缩放,大量溶剂校正和TLS约束。使用程序TLSMD 生成Gankyrin的5个TLS组和scFv F5的4个组。各个B原子因子各向同性地精制。然后根据电子密度图中未指定的峰放置溶剂分子。在Gankyrin-scFv F5复合物中,在2.5Å处精制而没有σ截止值,最终模型包含针对Gankyrin的224个残基(3-226),针对抗体scFv F5的225个残基(3-119和134-244),以及428个水分子。如前所述计算游离R值。结构图是使用PyMOL程序生成的
参考资料
- J. Mol. Biol. (2014) 426, 3729–3743. 《Restricted Diversity of Antigen Binding Residues of Antibodies Revealed by Computational Alanine Scanning of 227 Antibody–Antigen Complexes》
