【4.4.5】预测人类人群中的HLA CD4免疫原性(CD4episcore)
官网:http://tools.iedb.org/CD4episcore
背景:从对T细胞应答机制的基本理解和实际应用方面,对T细胞免疫原性的预测都是相当感兴趣的话题。HLA结合亲和力通常用于预测T细胞表位,因为HLA结合亲和力是人类T细胞免疫原性的关键条件。但是,由于高水平的HLA分子变异性,HLA以外的其他潜在因素以及HLA分型数据的频繁缺乏,使人群的免疫原性变得复杂。为了克服这些问题,我们探索了另一种方法来鉴定能够区分免疫原性肽和未识别肽的共同特征。
方法:将来自同行评议的发表论文的优势表位集与来自相同实验/供体的阴性肽结合使用,以训练神经网络并产生“免疫原性评分”。我们还将免疫原性评分的性能与先前描述的基于人群水平上HLA II类结合的免疫原性预测方法进行了比较。
结果:免疫原性评分在一系列独立的数据集中进行了验证,这些数据集来自已发表的文献,代表了57项独立研究,其中通过测试跨越不同抗原的重叠肽段评估了人群的免疫原性。总体而言,这些测试数据集对应于2,000多种肽,并在1,600多种不同的人类供体中进行了测试。 7等位基因方法的预测和免疫原性评分与相似的性能相关联[ROC曲线下的平均面积(AUC)值分别为0.703和0.702],而联合方法的平均AUC为0.725。平均AUC值的增加与免疫原性评分相比(p = 0.0135)显着,与7等位基因方法相比(p = 0.0938),观察到显着的趋势。现在可以使用Immune Epitope Database网站( http://tools.iedb.org/CD4episcore )上的CD4 T细胞免疫原性预测工具免费获得这种新的免疫原性评分方法。
结论:新的免疫原性评分可从蛋白质序列开始,且无需HLA分型,可在人群水平上预测CD4 T细胞的免疫原性。它的功效已在不同抗原来源,种族和用于表位鉴定的不同技术的背景下得到验证。
一、前言
T细胞表位的鉴定在从疫苗设计到癌症,过敏和传染病领域的诊断等多种免疫学背景中具有重要意义。目前,大多数表位鉴定是使用生物信息学预测系统进行的,该系统旨在鉴定T细胞的免疫原性以及解剖T细胞应答发展的潜在机制。当前,大多数T细胞预测方法都是基于HLA结合亲和力的预测,而HLA结合亲和力是人类T细胞免疫原性的关键条件。但是,缺乏能够在人群水平上预测免疫原性的有效策略,当没有HLA分型数据时,这一点尤为重要。为克服此问题,重要的是要确定能够区分免疫原性肽和未识别肽的常见HLA结合亲和力特征。 HLA分子的两个主要类别在免疫学背景中很重要。 I类分子向CD8 T细胞呈递表位,而II类分子向CD4 T细胞呈递表位。 HRO I类结合的预测已经达到了很高的准确性,并且ROC曲线下的面积(AUC)值大于0.9,类似地,HLA II类预测在最近几年也有了显着改善,达到了显着的准确性水平(使用AUC值在0.760-0.870之间。但是,HLA分子具有高度的多态性,在种群水平进行表位预测必须考虑到这种高度的异质性。
我们以前曾证明,在HLA I类的情况下,关注25-30个主要的HLA A和B等位基因变异体可以覆盖很大一部分普通人群(11)。同样,对于HLA II类,大约40–50个等位基因变体覆盖了最常见的等位基因变体(12)。 HLA结合的预测通常使用等位基因特异的算法进行,因为不同HLA的结合基序相当多样。但是,在HLA II类的情况下,还应注意,不同变体的表位结合之间存在高度重叠(13)。实际上,已经表明,显性识别的表位通常能够结合许多不同的HLA II类等位基因。这些抗原决定簇(称为混杂抗原决定簇)在总体水平上占总应答的50%或更多(14)。
“ 7等位基因方法”是基于混杂表位的预测而专门针对人群水平(15)预测HLA II类反应而优化的。尽管该方法具有显着的预测价值,但也可以预期,许多预测或实验证明结合II类HLA分子的肽可能不会诱导T细胞应答。这是因为尽管HLA结合是必需的,但其本身不足以实现T细胞的免疫原性。其他因素,例如抗原加工和能够识别任何给定的MHC /表位复合物的TCR库大小,是最终确定免疫原性的关键因素(16-18)。
特别是,已经显示出TCR库是塑造表位免疫优势的关键因素(19-23)。对于HLA I类,已设计出不同的算法来评估某种肽序列中某些氨基酸的存在,这些氨基酸可能与TCR相互作用,从而成为表位固有免疫原性的潜在因素(24-26)。
在本研究中,我们评估了一种方法,该方法通过训练神经网络(NN)与在一般人群中占优势的免疫原性表位结合,来在人群水平上预测HLA II类免疫原性,而与特定的HLA单倍型无关。因此,该方法不仅可以探测HLA结合的影响,而且还可以潜在地检测将在潜在表位的一级序列中编码的HLA II类结合以外的因素。
二、材料和方法
2.1 数据集
用于训练的数据集完全来自我们实验室中使用一致技术作为依赖严格控制的数据集的手段生成的实验数据。此外,我们还将与阳性四聚体数据相关的表位作为训练的一部分,因为在分析T细胞反应时,四聚体数据被视为质量和特异性的“金标准”。相反,用于验证的数据集是使用广泛的技术和抗原从科学文献中得出的,并且是从世界各地的不同实验室得出的。进行此选择是为了确保所提供验证的可靠性。
2.1.1 培训数据集汇编 Training Dataset Assembly
我们使用了15-mer肽,这些肽来自于同行评审文章中描述的几个数据集,或通过采用相同实验方法的内部研究获得(表1)。在某些情况下,根据中期分析选择了抗原决定簇,这些抗原决定簇与发表的文章中的最终抗原决定簇清单不完全匹配。通过ELISPOT分析针对以下一种细胞因子:IFNγ,IL-5,IL-17或IL-10在5–150个供体的队列中测试了该肽的免疫识别。这些表位的完整列表在补充材料的表S1中描述。在这项研究中,总共选择了1,032个表位作为阳性。根据特定标准从表1中列出的相同数据集中选择负肽:肽在所有测试中均应为阴性,仅包括来自至少识别出一种正肽的蛋白质的肽。此外,从数据集中删除了多次测试的任何肽(由于多项研究测试了同一生物的抗原/过敏原),从而对同一供体产生了相反的反应。总的来说,获得了5,739个阴性肽(补充材料中的表S2)。在某些情况下,出于技术原因,有必要对标准中的特定设置进行调整,如下所述。



结核分枝杆菌(TB)抗原
我们从疫苗候选抗原中选择了65个先前已知的15-mer表位,它们捕获了80%的应答(27-29)。
蒂莫西·格拉斯(TG)已知过敏原 Timothy Grass (TG) Known Allergens
先前的研究确定了20个表位,这些表位占TG过敏个体对一组TG来源的花粉抗原(Phl p过敏原)总反应的79.5%(14、31、32)。大多数数据集由15个单体组成,因为它们基于HLA II类结合预测(15、99)。但是,由于其中一些表位不是15-mers,为了与其他数据集进行比较,将较长的表位分解为15-mer,组成较长肽的每个15-mer被归为阳性,用于阴性肽的过程相同。另外,描述了19种肽覆盖NTGAp19肽库,选择这些肽以涵盖针对所筛选的所有NTGA肽的总IL-5反应的至少40%(30)。
屋尘螨(HDM)过敏原 House Dust Mite (HDM) Allergens
肽组包括34个最主要的表位,累计占我们筛选中检测到的总过敏原特异性反应的90%(32)。与TG类似,更长的区域被解构成15聚体,总共产生52个肽。
蟑螂(CR)过敏原 Cockroach (CR) Allergens
基于大于1,000的总斑点形成细胞(SFC)值选择了71个最主要的表位(33)。
登革热(DENV)抗原 Dengue (DENV) Antigens
在约10个HLA匹配的供体中测试了预测与各种常见DRB1等位基因结合的肽。 该组包括325个表位,在至少两个供体中PBMC呈阳性,该PBMC来自来自科伦坡(斯里兰卡)地区的正常供血者,对DENV抗体呈血清反应阳性,因此代表自然感染(34)。 负肽是在至少10个供体中测试的肽,被发现均为一致阴性。
促红细胞生成素 Erythropoietin
Tangri等。 筛选了重叠的肽,并报告了至少40%的供体可以识别的9个表位(35)。
CRJ1和CRJ2日本雪松过敏原
该组包含重叠的15-mer,跨越CRJ1和CRJ2变应原(36)。 我们基于两组过敏性供体中的任何一个的平均响应强度> 100 SFC(IL-5和IFNγ的总和),选择了30个显性表位:在日本居住时间较长的那些和未在美国居住的美国致敏的供体 在日本。 总共18个阴性对照肽源于过敏原CRJ1和CRJ2,并根据一个或多个供体的响应频率和单个SFC响应<100 SFC进行选择。
小鼠过敏原
在7个等位基因算法中,主要从小鼠过敏原衍生的肽已在22个供体中进行了测试(37)。 根据总SFC> 150定义了总共89个显性表位,并在至少两个供体中被识别。
新型屋尘螨抗原
用7等位基因方法从20个HDM过敏供体中的96种HDM(新颖和已知)蛋白中预测了筛选出的肽(38)。我们选择了106个优势显着的表位,这些表位在多个供体中被识别,总体大小总计> 300 SFC(约占总响应的50%)。
百日咳疫苗抗原
肽组由覆盖八个抗原残基的16-聚体组成,涵盖了抗原的整个序列。我们选择了在分析的53个供体中的至少4个中识别的前100个表位,约占总应答的75%(39)。
豚草过敏原
该组包括由八个氨基酸重叠的16-聚体,涵盖了抗原的整个序列(40)。总共选择了15个表位,占总反应的75%。如果存在变体,则选择最常见的变体。
破伤风类毒素(TT)抗原
我们选择了一组28个表位,它们在至少20个供体中有2个被识别(41),并通过7等位基因方法预测(15)。作为对照,我们从免疫表位数据库(IEDB, www.iedb.org )或Antunes等人的研究中选择了一组57种已被研究但未被识别的肽。 (41),以及该方法未预测的另外一组41个肽,Antunes等人的研究也未识别。在IEDB中也未将其确定为积极的人类反应。在第三组41个肽的情况下,破伤风组中有261个15聚体。其中有124个被预测是7个等位基因中位数百分等级≤20.0的结合物。从137个非预测肽中,选择7个等位基因中位数百分位数等级> 40.0的肽(67个肽)进行筛选,以纳入非预测AND非表位组。从该列表中,我们消除了与任何表位重叠(超过5个AA残基)的肽(在我们的研究中被确认或在IEDB中标注为阳性)。其余的41种肽被包括在Antunes等人未曾预测和识别的“对照肽”组中。研究也未在IEDB中确定为正面反应。
ZIKA病毒(ZIKV)抗原
用14天的再刺激方案在18个供体中测试了跨越ZIKV蛋白质组整个序列的一组15-mer肽。 在至少两个供体中总共定义了48个表位为阳性(Grifoni等人,未发表)。
黄热病(YF)抗原
测试的表位组包括94种先前描述的具有已知HLA II类限制(IEDB)的YF CD4 T细胞表位,以及预计结合不同HLA DRB1分子的肽组。 将来自42个接种YF17疫苗的供体的CD4 + T细胞与自体抗原呈递细胞和HLA匹配的YF DRB1预测肽共培养。 14天后,如先前所述确定针对单个肽的IFNγ反应(100)。 表位定义为引发SFC为664 SFC / 106或更高的肽。 这导致鉴定出42种独特的肽(Weiskopf等,未公开)。
2.1.2 IEDB验证数据集
为了生成其他数据集以评估各种预测方案的性能,我们试图确定报道重叠肽研究的文献记录。因此,我们向IEDB询问了包含与该类HLA II类限制性T细胞有关的阳性和阴性记录的论文。该查询确定了870篇论文;通过摘要中提到的“重叠”对这些内容进行了过滤,从而对其进行了进一步细化,从而产生了183条记录。
手动检查了这些记录的摘要,以选择与重叠肽组的免疫原性研究真正相关的论文。在这一阶段,我们排除了与Phl p,TT,TB有关的记录(已在先前的文献中代表过)和基于转基因小鼠的研究,以获得102篇相关论文。
接下来,我们删除了肽大小小于15或研究供体少于10个的论文(共82篇论文)。这82篇论文中的每篇均进行了手动检查,由于各种原因,其他文件在进行手动检查时都被丢弃了,包括该论文未报告对整套重叠肽段的测试,对报告的阴性结果或肽段大小的报告不明确,对阳性结果之间没有明显的区分。和阴性反应,没有解卷积的肽库测试以及类似问题。
最终选择了57篇论文(表1)。对于每篇论文,根据公开的数据和作者的解释,我们捕获了占主导地位的抗原决定簇,这些抗原决定了大多数回复和/或在多个捐赠者中始终为阳性。我们选择始终为阴性的肽作为相应的阴性对照。在测试大量供体且基本上所有肽均为阳性的研究中,我们选择了一个或多个供体中的肽阳性。补充材料的表S3A中提供了PUBMED ID的列表以及用于选择“顶部”表位和“底部”阴性对照的标准。补充材料的表S3B中提供了阳性和阴性对照肽的列表。
2.1.3 四聚体训练数据集 Tetramer Training Dataset
使用以下选择标准从IEDB(2015年6月访问)(101)下载了与在四聚体染色实验中描述为阳性的表位相对应的数据集:“仅阳性测定,表位结构:线性序列,T细胞测定:定性结合/多聚体/四聚体(四聚体),无B细胞测定,无MHC配体测定,MHC限制类型:II类,宿主生物:智人(人类)(ID:9606,人类)。”过滤导出的数据集,仅保留可获得源抗原蛋白ID的15-mer表位。对于每个独特的阳性肽,我们使用抗原基因组ID获取其源蛋白序列,并扫描该蛋白中所有可能的15个重叠10个氨基酸的聚体。然后将原始的阳性肽视为一种免疫原性肽,并将其余的获得的肽用作阴性肽。四聚体数据集具有124个独特的正值和5,319个负值,如补充材料的表S4所示。
2.2 NNAlign方法的基于人工神经网络(ANN)的预测
使用NNAlign方法对肽序列进行NN训练。该方法使用分类的肽数据进行训练,并识别嵌套的较短序列模式,这些模式组成了一个信息基序,可以将阳性和阴性实例分开。作为NNAlign的输入,我们使用了15-mer肽的序列及其指定的观察到的免疫原性评分(免疫原性1.0和非免疫原性0.0)。使用广泛的交叉验证对方法进行了训练,其中部分数据被排除在训练过程之外,仅用于评估目的。对于每种肽,该方法返回的预测分数在0.0到1.0之间,其中高值标识更多的免疫原性肽,低值标识非免疫原性肽。 NNAlign-1.4软件包是从 http://www.cbs.dtu.dk/services/NNAlign/ 下载的。对每种可能的基序长度从1到15进行了训练的方法都进行了交叉验证。交叉验证的数据是根据肽中的通用基元进行拆分的,最大重叠量为9,并改变了基元长度。使用稀疏和BLOSUM方案编码输入肽。没有对输入数据进行重新缩放。我们还选择保留原始数据中的重复侧翼,并且不使用偏移量重新对齐网络。使用每个网络体系结构的10个种子,使用5个隐藏的神经元对方法进行了训练。其他编码方法,NN设计的选择或其他学习算法的选择都有可能带来更好的结果,但是这种比较超出了我们当前手稿的范围。
2.3 接收器工作特性(ROC)曲线和AUC值
为了测量不同的方法如何将肽分为表位和非表位,使用了ROC曲线(103)。 根据预测分数的截止值,将肽分为免疫原性和非免疫原性,并获得真阳性(TPs)和假阳性(FPs)的数量。 通过绘制每个截止点的TP速率与FP速率的函数来绘制ROC曲线。 AUC是用于评估预测方法的预测性能的有用措施。 AUC值的范围从0.5到1,其中0.5对应于随机预测,1对应于完美预测。 可以将AUC值解释为随机选择的免疫原性肽的预测得分高于随机选择的非免疫原性肽的得分的概率。
2.4 HLA绑定预测
我们利用先前描述的7等位基因方法(15)得出HLA结合倾向。 7等位基因方法基于七个等位基因的中位数百分位数预测结合来预测免疫原性,这七个等位基因代表了普通人群中最常见的结合基序,可在IEDB网站上找到(104)。
2.5 生成两样本徽标
从15个肽段(表位和非表位)组合的所有数据集中创建了15个肽段的两个样本徽标(如果肽段较长,则从N末端提取15个残基)。对于两样本徽标,表位和非表位数据集(以FASTA格式的文件)均以默认设置提交到在线工具( http://www.twosamplelogo.org/cgi-bin/tsl/tsl.cgi ),但默认设置除外的p值设置为0.01,分辨率为600 dpi(105)。
2.6 统计分析
使用Prism 7(Graph-Pad Software,圣地亚哥,加利福尼亚,美国)进行统计分析。利用Pratt方法进行的非参数Wilcoxson配对配对有序秩检验用于评估不同AUC值集之间的显着性差异。
三、结果
3.1 人工神经网络衍生的免疫原性评分的推导和验证
我们根据实验室中不同的先前发表的多肽筛选研究组装了T细胞表位数据集(表1)。在所有情况下,均使用ELISPOT分析法筛选多肽,以检测哪些肽刺激了细胞因子的分泌。表1总结了针对每种肽组筛选的供体数,以及选择的肽与特定抗原重叠还是根据预测的结合亲和力进行选择。如“材料和方法”部分中更详细描述的,占大多数T细胞应答的优势表位被视为阳性(补充材料中的表S1,N = 1,032肽)。在任何供体中均未产生任何应答但来自至少一个肽为阳性的蛋白质的肽被视为阴性(补充材料中的表S2,N = 5,739个肽)。使用此否定分类的附加标准来确保识别的缺乏不仅仅是由于缺乏抗原呈递所必需的源蛋白。
该初始数据集用于训练一种基于NN的方法NNAlign(102)。 NNAlign方法采用未比对的肽组,旨在在肽内找到线性序列核心,从而将阳性(免疫原性)肽与阴性(非免疫原性)肽区分开。系统检索每个变异的序列核心长度,从单个残基到15个残基的免疫原性评分(范围为0-1),并使用五重交叉验证法评估了预测质量。几个序列核心长度显示的AUC值大于0.7,这通常被认为是良好的预测质量值,这表明ANNs根据肽基序显示出正肽和负肽之间的差异。就序列基序长度而言,交叉验证并未表明明确的最佳长度,因为对于3到12之间的基序长度,预测性能相似(图1)。 9个残基的基序长度与参与HLA和TCR的肽核心区的已知大小一致。因此,以下分析选择了9个主题长度。

3.2 结合免疫原性和HLA结合预测
为了同时考虑HLA结合和免疫原性预测(可能具有被TCR识别的能力),我们将基于ANN的免疫原性预测与HLA II类结合预测结合在一起。 仅描述了一种基于HLA在人群水平上的结合来预测表位的方法,即7等位基因法,该方法先前已根据免疫原性数据集进行了经验优化(15)。
为了结合免疫原性和HLA结合评分,我们使用7等位基因方法的中位数百分等级评分(HLA_score)(范围从0到100),并将其转换为百分位数后与基于NN的免疫原性评分相结合,因此 使用公式(Imm_score)=(1-神经网络免疫原性)×100,它的范围也可以从0到100,并且可以与HLA_score相当。两个得分的组合如下:
Combined score=α× Imm score+(1−α)×HLA score. (1)
接下来,我们在0≤α≤1的区间内系统地改变α的值。根据上式,当α= 1时,结果仅取决于NN的免疫原性预测,而对于α= 0时,仅HLA结合预测为用于定义免疫原性。
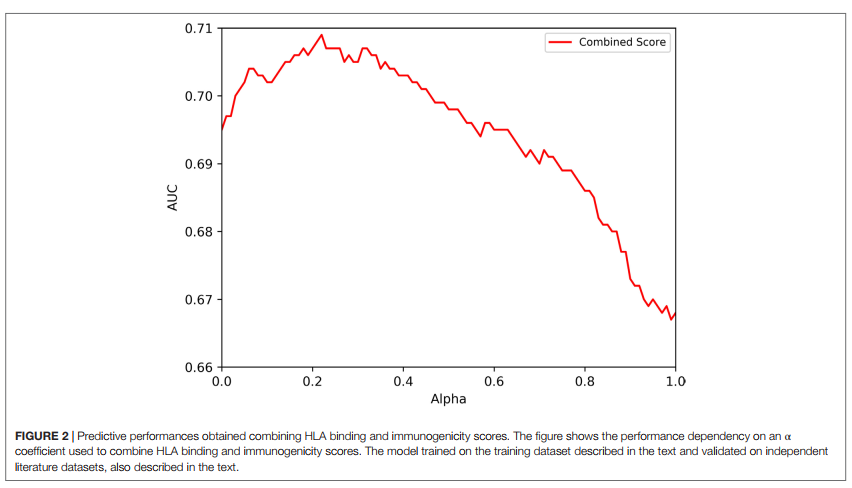
为了评估免疫原性评分,7等位基因方法及其组合的性能,我们使用了独立的基于文献的数据集。具体而言,我们在IEDB上搜索了描述测试与人类HLA II类限制性T细胞相关的重叠肽组的结果的论文。这些表位集因此代表了广泛的研究,代表了在全球科学界中进行的表位识别研究的“真实”肖像。这些表位集在补充材料的表S3A中列出,并在“材料和方法”部分中进行了详细描述,序列在补充材料的表S3B中提供。总体而言,对来自独立文献研究的总共57种不同组进行了整理,总共产生530个阳性和1758个阴性肽。图2描述了合并分数的预测性能,显示了为每个不同数据集获得的不同AUC值的平均值。 7等位基因方法的AUC值为0.695,免疫原性评分的平均AUC值为0.670。根据两种算法的组合,性能提高并在α值为0.50时达到0.71的峰值。
3.3 免疫原性评分的表现,消除了训练和测试数据集之间的冗余
预计包含其他数据点将提高NN模型的性能。因此,我们纳入了通过四聚体作图研究确定的CD4 T细胞表位的其他数据集。我们认为这将提供高质量的表位,因为四聚体染色测定法通常被视为表位表征的“金标准”测定法。通过查询IEDB获得通过四聚体染色测定法检测为阳性的15-mer肽,获得数据集。对于每个阳性肽,扫描其源蛋白以寻找重叠有10个氨基酸的15-mer肽,去除阳性肽序列,并将其余肽用于构建阴性数据集。最终的四聚体数据集由124个独特的阳性肽和5,319个独特的阴性肽组成(补充材料中的表S4)。
用于训练和评估NN模型的数据集包含一些冗余,这可能会影响评估并提高性能。为避免此问题,我们通过过滤掉共有9个mer序列的任何肽段,消除了训练集(表1和四聚体集的组合)和57个独立研究的验证集(表1)之间的任何冗余。
在进行的分析中,未观察到明确的最佳α。图2中的数据似乎表明最佳α值在0.2-0.3左右,而图3中的分析表明在0.4和0.6处有两个最佳峰。由于图3中的数据由于使用大量数据点进行训练而在本质上更加可靠,因此我们根据经验选择0.4作为要包括在下一组分析中的alpha。进行此分析时,7等位基因方法的预测和免疫原性评分与相似的性能相关(平均AUC值分别为0.703和0.702),而组合的方法再次提供了性能提升,达到0.725的平均AUC(图3)。与使用Wilcoxon配对配对有序秩检验的ap值为0.0135的免疫原性方法的平均AUC值相比,组合方法的平均AUC值显着增加,与7等位基因相比,显着的趋势显着ap值为0.0938的方法。这些结果以及通过四聚体数据集获得的结果证实,7等位基因和免疫原性评分方法本身具有显着的预测价值,在两种情况下,它们的组合均可增强其预测价值。

3.4 一般免疫原性图案的两样本徽标
接下来,我们分析了来自所有数据集的表位和非表位,以保留其位置残基,并使用来自肽N端的15个残基绘制了两个样本的徽标(105)(图4)。两个样本的徽标代表基于t检验计算的p值(<0.01)的抗原决定簇和非表位显着不同的氨基酸。在表位数据集中富集的氨基酸残基大部分带正电,而在表位数据集中消耗掉(并在非表位数据集中富集)的氨基酸大部分带负电。换句话说,表位在第9和11-14位上具有较高数量的带正电荷的残基,如精氨酸(R)或赖氨酸(K),而非表位在7-9和11-13位上含有天冬氨酸(D)和谷氨酸(E)。 。在非表位中还观察到对疏水残基的偏好[例如脯氨酸(P)和丙氨酸(A)],而异亮氨酸(I),苯丙氨酸(F)和天冬酰胺(N)在表位集中富集。为了进一步解决徽标的重要性,我们将数据集分为五组,每组包含总数据集的80%,补充材料中的图S1中的结果确认徽标显示的最普遍特征与使用整个数据集创建的两个样本徽标(图4)。这些结果表明,这些偏爱中的一些可能有助于T细胞识别或MHC结合,或代表加工酶的结果。这些可能性将在以后的研究中讨论。

3.5 表位预测阈值和在线工具的实现
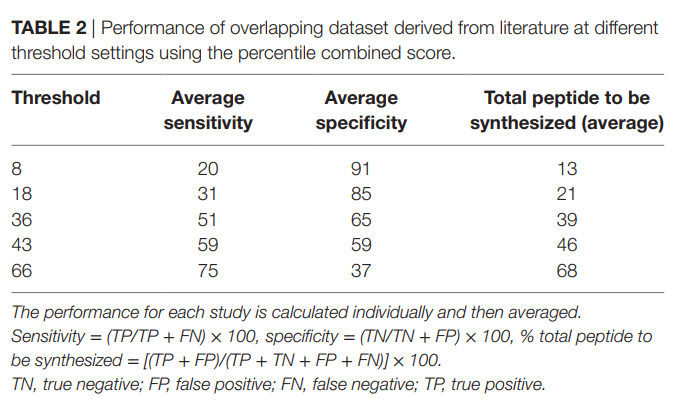
接下来,我们为每个研究使用从0到100的不同截止值(表2)来确定综合得分的表现。为此,我们使用α= 0.4时的百分组合得分计算了在不同阈值设置下从文献中得出的重叠数据集的性能。第一步,对于每项研究,我们计算以下数目:真实阴性(TN)定义为预测为非免疫原性的非免疫原性肽,FP定义为预测为免疫原性的非免疫原性肽,假阴性(FN)定义为免疫原性预测为非免疫原性的肽,而TP定义为预测为免疫原性的免疫原性肽。根据这些值,我们计算出灵敏度[=(TP / TP + FN)×100]和特异性[=(TN / TN + FP)×100]。最后,我们确定8、36和66的临界值允许分别捕获20、51和75%的表位,对应的特异性为91、65和37%。我们还使用以下公式估算了观察到的表位定义分数所需测试的肽分数:[(TP + FP)/(TP + TN + FP + FN)]×100。关联值为43具有相同的敏感性和特异性(59)。为了使该方法易于使用,我们还实现了该算法的在线版本(图5)。该工具可在IEDB网站上免费获得,网址为 http://tools.iedb.org/CD4episcore/。
四、讨论区
用于鉴定T细胞表位的生物信息学预测常用于设计和测试针对传染病,过敏和癌症的疫苗和诊断方法。虽然已经描述了几种基于HLA等位基因的特定预测算法(10)和基于MHC II类结合的T细胞表位预测策略(106-108),但仍缺乏有效的策略来预测人群水平的免疫原性,因此仍然具有重要意义利益。这很重要,因为在实际应用中,最经常遇到的HLA类型数据通常不可用。
在这里,我们报告一种方法,以识别区别于未被识别的HLA II类等位基因的,被CD4 + T细胞识别的免疫原性肽与非识别肽的序列基序。我们确认,先前描述的7等位基因方法(15)可有效预测表位,并且可以缩小用于生物学测试的肽段范围。重要的是,我们发现联合的HLA结合+免疫原性方法比单独的免疫原性预测显着改善,并且有明显的趋势表明联合的HLA结合+免疫原性方法比单独的HLA结合预测具有重要性。
我们应用了机器学习算法(NNalign),以识别特定长度的序列基序,以区分肽集-在我们的情况下是免疫原性肽和非免疫原性肽。我们发现,在8至11个残基之间的基序长度在不同数据集的分类中提供了最佳性能。该基序长度与与T细胞受体接触的表位残基以及HLA II类分子的表位核心结合特征的长度相符,后者也约9个氨基酸长(20、109 )。
与以前的研究一致,仅对HLA结合进行的预测增加很少,这一事实表明,HLA结合是塑造T细胞抗原决定簇的主要力量。但是,如其他研究之前所建议的那样,这种相对较小的增加也可能与HLA结合与抗原加工和TCR识别之间的协调进化有关(110)。
由于该方法仅基于免疫原性结果得出,因此此处定义的基序不仅可能与HLA结合有关,而且还可能包含针对TCR残基接触的总体偏好。但是,鉴于其衍生的无偏性,不能排除该方法也可能反映出完全不同的过程,例如通过HLA-DM进行调制或相对于亲和力提高HLA结合稳定性是该基序的实际来源( 111)。
非常短的基序(3、4个残基)的预测能力非常惊人。潜在的结构或机制基础可能反映了短片段的残基的显性影响,这些残基掺入了与HLA结合的显性残基,并紧靠在TCR识别中也显性的残基(15)。检查基序中的残基表明,在基序的中间避免了氨基酸侧链较小的肽,而具有较长侧链的残基却被过多代表。这在质量上类似于我们先前对HLA I类限制性表位的发现,并且在使用单残基取代的实验研究中已有报道(112、113)。这进一步支持了所鉴定的基序与更可能与TCR结合的肽的性质相吻合。在靠近N端的位置富集F,M,L至少部分对应于MHC-II的P1锚点,后者在几个基因座和等位基因变体中具有相似的特异性。
我们的结果已经过扩展的数据训练,这些数据来自不同的方法和不同种族的人群,并与传染病,过敏和自身免疫性有关。尽管偏向某些HLA等位基因并且可能在阴性组中包含许多表位(即来自相同蛋白质的其他表位而不是所考虑的四聚体),但经过四聚体训练的算法似乎表现更好。我们推测这可能是由于四聚体表位代表通常占优势的表位这一事实,而表位又被证明与混杂的HLA结合物相对应。总体而言,组合的训练集对应于来自300多种不同抗原的14000多种肽,并在2500多种不同的人类供体中进行了测试。我们认为,这是我们研究的重要方面,因为它可以确保我们的构建模型(与7等位基因方法,免疫原性评分和联合方法有关)有效,而与抗原来源,不同种族和不同技术无关表位鉴定。我们的预测方法对于产生现成的病原体或常见肿瘤标志物疫苗肽库可能有用。相反,在HLA型个体中进行NGS测序后,该方法可用于最佳选择覆盖个体化肿瘤衍生新表位的肽。
该算法可在IEDB网站(101)上找到,并且我们估计,结合使用免疫原性评分和7等位基因方法,可以合成24%可能重叠的15-mers来捕获50%的总表位。这将转化为具有72个15-mer肽的300个残基蛋白质的覆盖范围。 T细胞表位预测的未来改进可能受益于从HLA II类分子洗脱的肽的大规模数据集,识别表位的特定TCR的数据集以及揭示MHC II类加工途径中介体作用的数据集的增加的可用性-DM。
即使采用这种方法,AUC值也低于MHC-I分析(1)。但是,应记住,这些AUC值是指在包含具有多种限制的T细胞的人群水平上的预测,而MHC-1的较高AUC值通常是指等位基因特异性的预测。但是,从MHC-II到MHC-I的当前方法的应用面临特定的挑战。在MHC-1中,人们认为有更多的HLA特异性表位选择,这与当前方法的直接应用存在争议,但是alpha分析有可能识别出任何不依赖HLA的成分。最后,将有兴趣开发一种类似的方法来开发HLA I类抗原决定簇的HLA不可知预测因子。最近的数据表明,有可能凭经验开发HLA I类抗原表位“巨池”,该种群可覆盖普通人群,而不论其种族如何(114,115)。未来的研究将集中在类似的方法上,用于HLA不可知类I类受限表位的预测。
参考资料
- Dhanda et. al.: Prediction of HLA CD4 immunogenicity in human populations, Frontiers in Immunology, 2018, 9, 1369. https://www.frontiersin.org/articles/10.3389/fimmu.2018.01369/full
