【4.4.4.5】预测TCE(MHCI和MHCII)--Rankpep
官网: http://imed.med.ucm.es/Tools/rankpep.html
T细胞免疫应答是由抗原表位驱动的,因此其识别对于了解疾病的发病机制和病因以及疫苗设计非常重要。有两种类型的T细胞抗原决定簇,分别称为CD8和CD4,它们仅在MHCI和MHCII分子的上下文中被相应的T细胞类型识别。参与两组T细胞对于增强针对癌细胞和病原体的防御性免疫反应是理想的。因此,我们已经开发了该位点,用于预测肽与MHCI和MHCII分子的结合,以及随后对CD8-和CD4-T细胞表位的预期。抗原肽与相关MHC分子结合之前必须进行适当的加工。顺便提一句,大多数MHCI限制的抗原决定簇(CD8-T细胞抗原决定簇)的C端是由蛋白酶体切割产生的,因此,蛋白酶体特异性对于确定T细胞抗原决定簇很重要。因此,该位点还可以确定预测的MHCI配体的C端是否是蛋白酶体切割的结果。 RANKPEP Web服务器中还实现了可变性屏蔽功能,以专注于保守表位的预测,因此可以帮助避免因突变而导致的免疫逃逸。
二、Rankpep:MHC-肽结合预测
与给定的MHC分子结合的肽具有序列相似性。毫不奇怪,传统上将序列模式(sequence patterns)用于预测与MHC分子结合的肽。但是,这种序列模式已被证明过于简单,因为结合基序的复杂性无法通过模式[1]中存在的少数残基精确地表示出来。为了克服此限制,RANKPEP使用位置特异性评分矩阵(PSSM)或已知与给定MHC分子结合的比对肽组中的谱作为MHC肽结合的预测因子。
2.1 PSSM用于预测MHC肽的结合
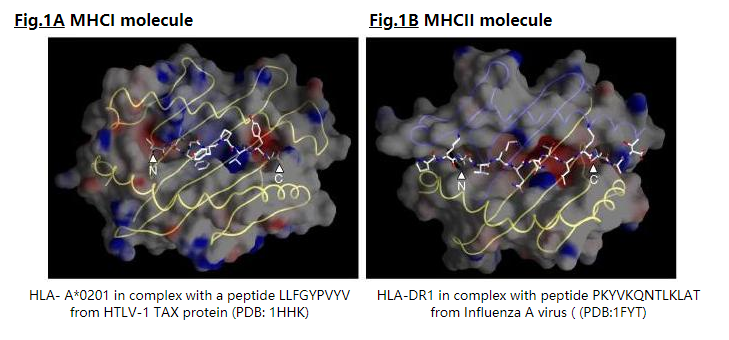
为了使谱(profile )成为结合基序的良好描述,必须通过结构和/或序列相似性对肽进行比对[2]。 MHCI和MHCII分子以相似但不同的模式结合肽(图1),获得的MHCI-和MHCII-配体的比对与该肽与其MHC类的结合模式一致。
-
MHCI配体的长度短(8-11),因为它们被限制在MHCI肽结合槽中,其N和C末端通过氢键网络连接到MHCI分子的保守残基上(图1A) 。因此,与相同的MHCI结合的肽可以相差一个或两个氨基酸,如中所述,如果这些肽的长度相同,则可以更好地保证这些肽的正确结构比对。因此,我们已经将与给定的MHCI分子结合的肽分离为仅包含相同长度肽的子集,并从无缺口的嵌段比对中创建了独立的PSSM。
-
MHCII分子的肽结合槽是开放的,以N和C端都可以延伸超出结合槽的方式结合肽(图1B),因此,与MHCII分子结合的肽的长度变化很大(9-22)。然而,只有9个残基的肽核心适合提供结合能的MHCII结合槽。 MHCII配体之间不良的氨基酸序列相似性以及它们在序列长度上的巨大变异性使其难以对齐。因此,对于MHCII配体的比对,我们使用了基序发现程序MEME(6),包括与MHCII-肽结合方式一致的先验信息:
- 每个MHCII配体只有一个结合核心;
- 所有肽序列定义相同的基序,
- 基序的长度为9。
使用PROFILEWEIGTH(对于MHCI-配体)从MHC-配体的这些比对中获得PSSM,它们使用分支比例序列权重或BLK2PSSM(基于位置的权重)(对于MHCII配体)。
2.2 使用PSSM对MHCI肽结合进行评分
任何肽序列(查询)与给定MHCI的结合潜力(得分)是通过将相关PSSM与蛋白质片段进行比对,然后加上与残基类型和位置相对应的图谱得分而获得的。 为了使用PSSM搜索蛋白质序列中的MHC配体,我们使用Python编写的动态算法,该算法会对PSSM宽度长度的所有蛋白质片段进行评分,并对其进行相应排序。 评分从每个序列的开头开始,PSSM每次在序列上滑动一个残基,直到序列结束。 此外,为了从排名的肽列表中缩小潜在的结合物,我们将结合阈值定义为评分值,该评分值包括PSSM中90%的肽。 该结合阈值内置于我们的每个矩阵中,描述了得分最高的肽中推定的结合剂(binders )的范围。
2.3 PSSM预测MHC肽结合的表现
如果PSSM是MHC肽结合的良好预测因子,则应从其蛋白质来源中的高得分肽中预期MHC限制性T细胞表位。 在此假设下,我们发现可预测约80%的MHCI限制表位,即最高得分肽的2%阈值。 但是,在〜5%的最高得分肽中发现了〜80%的MHCII限制性表位。 因此,与MHCI限制的表位相比,使用这些PSSM正确识别MHCII限制的表位需要大量的预测肽 铁汉 16:24:48
三、Rankpep:分裂预测 Rankpep: Cleavage Predictions
T细胞表位的预期很大程度上取决于MHC肽结合的预测。 在结合MHC之前,必须进行正确的肽加工。
- MHCII限制性表位的加工发生在内体区室中,它是由几种内肽酶与氨基和羧基肽酶联合介导的(10、11)。 这种复杂性使得鉴定与II类限制性肽的加工有关的任何模式变得困难。
- 另一方面,有实验证据表明,MHCI限制的抗原决定簇的C端仅由蛋白酶体介导的胞质蛋白的蛋白水解产生(12)。 因此,蛋白酶体在确定CTL表位中起着至关重要的作用,可以使用统计语言模型根据MHCI限制肽及其C端侧翼区域对它的特异性进行建模。
3.1 方法与实现
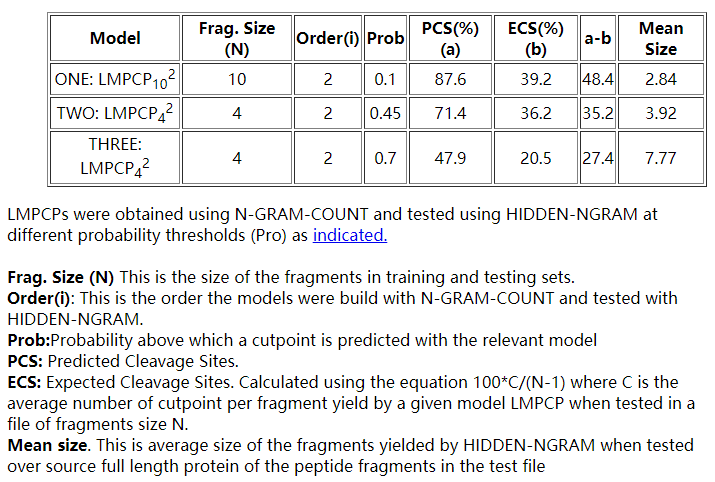
蛋白酶体的切割发生在蛋白质内的优先位点( preferential sites)上,蛋白酶体加工的抗原肽的序列信号在切割位点的P1(抗原肽的C末端)及其紧邻的P1’残基特别保守(13)。蛋白酶体切割的预测类似于语言标记(建模语法标签的位置,如标点符号)的问题,因此我们使用SRI语言建模工具包(SRILM)(14)进行蛋白酶体切割位点的统计建模。从包含人MHCI分子限制的332种抗原的C末端和侧翼区域的数据库中获得用于蛋白酶体切割统计模型的训练集。蛋白酶体切割预测的语言模型(LMPCP)是根据来自上述数据库的可变片段长度的肽片段训练集创建的。使用HIDDEN-NGRAM,对LMPCP在不同的切点概率(0.35至0.70)下,对训练集中未包含的肽片段及其整个蛋白质来源的测试文件进行了测试。对于每个概率阈值,测试了23个不同的LMPCPNi(N = 10、6、4; i = 1 N-2),其中N是训练和测试集中片段的片段大小,而i是所测试LMPCP的顺序。最佳结果(预测的切割位点,PCS> 75%)是在软切割概率(0.35-0.5)下从LMPCP获得的,表明蛋白酶体特异性的本质比语法标记(grammar tagging)的刚性低得多
3.2 Models
RANKPEP中提供的蛋白酶体切割(LMPCP)的语言模型如下:
四、Rankpep:免疫优势过滤器 Rankpep: immunodominance filter
T细胞表位的免疫原性取决于几个因素:
- 对其肽的蛋白质来源进行适当且有效的加工;
- 与MHC分子稳定结合的肽;
- TCR识别与MHC-肽的能力。
为了准确预测T细胞表位,需要对这三个过程进行适当的计算建模。 到目前为止,在表位预测算法中仅考虑了肽与MHC的结合以及针对I类MHC限制的表位的处理。 但是,在此版本的Rankpep中,我们实现了TCR对肽进行免疫显性识别的模型。
4.1 方法与实现
通过比较一组免疫原性肽与一组非免疫原性肽,对可能使它们更容易被TCR识别的肽中潜在的免疫优势特征进行建模。为了最小化由于MHC结合和加工而产生的任何贡献,仅选择了与MHC I类分子的高亲和力肽结合剂,并且未考虑侧翼区域。
- 免疫原性肽组由101个具有高MHC结合和高T细胞活性的肽组成。
- 另一方面,非免疫原性组由〜400个具有高MHC结合力且无T细胞活性的肽组成。所有考虑的肽均为9聚体。
- 使用支持向量机对这两组差异进行建模,它们在残基组成,氨基酸序列和两者的残渣上均受这两组训练。
4.2 结果
经过序列[1],理化性质[2]或两者结合[1 + 2]训练的SVM分类器能够区分免疫原性肽和非免疫原性肽,但准确性较低。基于理化性质的分类器能够胜过仅基于序列的分类器,因此表明肽的性质可能在决定免疫优势中起关键作用。 在残基特性和氨基酸序列上进行训练的基于SVM的免疫主分类器能够区分免疫原性肽与非免疫原性肽,准确度为60.0%阈值0.5。较低的累积量可能是由于我们将所有肽汇集在一起,而不管MHCI限制如何。不幸的是,目前对于单个MHCI限制元件,没有足够的免疫原性和非免疫原性数据。
4.3 Models
在Rankpep中,我们选择了基于残基特性和氨基酸序列训练的基于SVM的分类器的免疫优势过滤器。 如果将此过滤器设置为“开”,则服务器仅返回那些被归类为免疫线蛋白的肽。 由于免疫优势分类取决于阈值,因此我们提供了三个可选阈值供您选择。
- 阈值1.0:灵敏度为49.5%,特异性为76.0%
- 阈值0.5:敏感度59.4%,特异性69.4%(默认)
- 阈值0.0:敏感度68.3%,特异性60.9%
由于分类方法的准确性低,我们建议仅将这些免疫原性过滤器用于大型基因组规模的表位预测,以限制潜在表位的数量。
五、Rankpep:序列变异性屏蔽 Rankpep: Sequence variability masking
我们已经注意到,突变为某些致病性生物体(例如HIV)利用了逃避免疫的手段。 响应于该限制,可以从多个序列比对中获得RANKPEP预测。 在这种情况下,服务器首先创建一个共识序列,在该共识序列中,将可变性位置掩盖,并且将绑定预测限制在没有可变位置的片段中。 如Reche和Reinherz(15)所述,使用与Shannon熵方程式(16)完全相同的变异性度量(V),从多个氨基酸序列比对中计算序列变异性
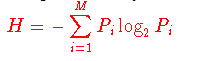
其中Pi是氨基酸类型i的残基分数,M等于氨基酸类型数20。 V的范围是0(总保守度,在该位置仅存在一种氨基酸类型)至4.322(在该位置均表示20种氨基酸)。 注意,为了获得最大值V = 4.3,至少需要20个序列。 间隙符号(-)被认为是用于推导共识序列的,但不会为变异性计算而计算。 在给定序列变异性阈值Vt的情况下,仅对那些以V <= Vt为最常见氨基酸的位置从序列比对中生成共有序列,而可变位位置(V> Vt)被掩盖并在共有序列中表示 带有“。” 符号。 在MHC肽结合的RANKPEP预测中不考虑带有被屏蔽位置的片段。
参考资料
