【4.4.4.3】SMM预测TCE
官网:http://tools.iedb.org/processing/
分子生物学中的许多过程涉及对核酸或氨基酸短序列的识别,例如免疫原性肽与主要组织相容性复合物(MHC)分子的结合。根据实验数据,可以构建这些过程的序列特异性模型,例如序列基序,评分矩阵或人工神经网络。这些模型的目的是双重的。
- 首先,他们可以提供实验结果的摘要,从而可以更深入地了解序列识别中涉及的机制。
- 其次,此类模型可用于预测尚未测试的序列的实验结果。
在过去,我们报告了一种生成这种模型的方法的发展,称为稳定矩阵法(SMM)。该方法已成功应用于预测肽与MHC分子的结合,与抗原呈递(TAP,antigen presentation)相关的转运蛋白的肽转运以及蛋白序列的蛋白酶体切割。
在此,我们将SMM算法的实现报告为可公开获得的软件包。讨论确定该方法最适合的问题类型的特定功能。该软件包的优势在于:(1)生成的输出易于解释;(2)输入和输出都是定量的;(3)内置了用于处理实验噪声的特定计算策略;(4)该算法旨在有效地处理有限的实验数据,(5)随机肽库和常规肽的实验数据可以轻松组合,并且(6)可以在序列位置之间整合成对相互作用。
使SMM方法公开可用,使生物信息学家和实验生物学家可以轻松访问它,将其性能与其他预测方法进行比较,并将其扩展到其他应用程序。
一、前言
每当收集实验数据来检查过程时,研究人员将尝试(隐式或显式)定义描述过程的模型。建立模型的目的是为了概括实验数据,从而可以预测新的实验结果,或者获得对实验过程的深入了解。在生物科学中,模型通常是隐式构建和口头表达的,例如“蛋白酶体优选在疏水性氨基酸后裂解”。这种模型易于理解,并且经常反映出可以从一些困难的实验中收集到的所有知识。但是,随着高通量生物实验的出现,基于大量数据生成更多定量的数学模型变得可行。为了建立模型,可以将进行实验的形式正式描述为收集成对的实验参数x和实验结果(测量值:ymeas)。建立模型等效于找到一个函数f(x)= ypred≈ymeas。在这里,我们将模型函数f称为预测工具,并使用实验观察来生成模型函数的训练集T =(x,ymeas)。
开发预测工具是一个两步过程。首先,必须选择一种能够生成特定工具的通用预测方法,例如某种类型的神经网络,分类树,隐马尔可夫模型或回归函数。这种选择在某种程度上取决于可用的实验数据。但是,只有少数几种预测方法显然不适用于某种类型或数量的数据,从而留下了几种可能合适的方法供您选择。在实践中,科学家的个人经验通常会决定他使用哪种预测方法,因为学习应用新方法通常比稍微好一点的模型的成本要高得多。
选择一种方法后,将其应用于实验数据以生成特定的预测工具。第二步通常使用几个不同的术语,例如“监督学习”,“拟合模型”或“回归”。每种方法都有自己的形式主义来描述如何完成,但是从本质上讲,一种方法能够生成一类特定的功能,从中选择一种,可以最大程度地减少训练集T的实测结果与预测实验结果之间的差异。
在这里,我们描述了一种实现SMM预测方法的计算机程序,该程序可用于对可量化生物过程的序列特异性进行建模。在这样的实验中,输入参数×对应于氨基酸或核酸序列,实验结果ymeas是测量过程效率的实数。该方法利用训练集,其中所检查的序列都具有相同的长度。
SMM方法先前已成功应用于MHC结合[1],TAP转运[2]和蛋白酶体切割[3]的预测。但是,它的软件实现没有公开可用,因为它依赖于具有限制性许可条款的商业库。而且,将该方法应用于新问题需要手动更改源代码,这对于外部用户而言是不可行的。这两个问题都在此处提供的软件实现中得到了解决。此外,该手稿还描述了两个特定功能,我们发现它们可以有效地生成高质量的模型,并且可以轻松地用于其他预测方法,即处理有限数据和组合肽库。
SMM软件并不适用于“经典”序列分析,可以将其粗略地定义为比对相关序列,以鉴定保守残基或生成可识别其他相关序列的分类器。 而是,典型的应用是序列识别事件的表征,例如蛋白酶的序列特异性切割效率。 该表征不假定不同识别序列之间的任何进化关系。 就科学领域而言,这使得SMM软件针对生物化学或分子生物学中的应用。
二、 Implementation
2.1 代码
SMM算法以C ++代码实现。 仅使用标准库或免费提供的外部库。 这两个外部库是处理XML输入和输出的Tinyxml [4],以及用于有效矢量和矩阵运算的Gnu科学库[5]。 已在Windows系统上使用Visual C ++并在Debian和Suse Linux系统上使用g ++(1.5或更高版本)对源代码进行了编译和测试。 Windows可执行文件也可用。 源代码,文档和示例可作为本手稿的其他文件1和2获得,也可以在项目主页上的 http://www.mhc-pathway.net/smm 上获得。
在运行Windows XP的标准2.6 GHz Pentium 4 PC上,从一组包含300个8-mer肽的典型训练数据中创建预测工具大约需要10分钟。
2.2 输入输出
该程序期望将XML文档作为其标准输入。对于计划的用户社区而言,更复杂的(图形)用户界面可能不会引起很大的兴趣,其中大多数用户可能会从脚本中调用可执行文件或将代码集成到现有程序中。 XML输入文档包含用于生成新工具的培训数据(图1)或应使用先前生成的工具进行预测的序列。程序的输出还是XML格式。对于所有类型的输入和输出,都提供了定义其确切结构的XML模式。模式包含每个输入元素的注释,记录其预期用途。对于最常见的应用,存在简化的替代输入格式,即从一组标准氨基酸序列生成评分矩阵。该程序提供了几个示例输入文件,这应使用户可以轻松地使用自己的数据创建类似的输入文件。
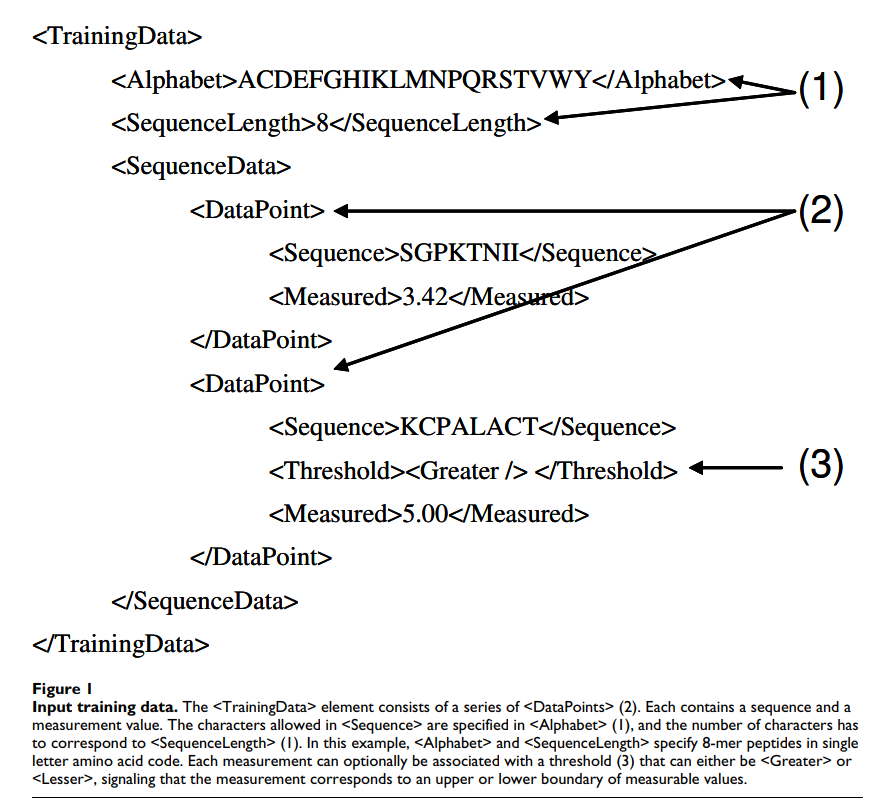
2.3 核心算法
氨基酸可以被编码为长度为20的二元向量,除编码特定氨基酸的一个氨基酸外,所有位置均为零。 扩展该表示法,可以将长度为N的肽编码为N * 20二元向量。 可以对核酸残基进行相同的转化,得到N * 4长度的载体。 遵循类似规则,可以将任何固定长度的序列转换为固定长度的二进制向量。 SMM算法期望这样的向量作为实验输入参数“ x”。
对于一组实验,可以将向量tx(其中tx表示转置的向量x)堆叠起来,得到矩阵H。图2显示了一组核酸序列的例子。矩阵H乘以一个向量 w为序列中每个位置的每个可能残基分配一个权重。 这将生成预测向量ypred


三、结果与讨论
以下各节描述了SMM方法的特定属性,并作为何时以及如何应用的指南。可以在以前的出版物中找到验证SMM算法并将其与其他预测方法进行比较的其他数据[1-3]。
3.1 线性模型
如果没有合并任何对系数,则SMM方法的输出向量w是标准的“得分矩阵”,该矩阵量化输入序列中每个位置上每个残基对预测的贡献。这样的矩阵易于解释和分析,而无需任何其他软件或如何生成矩阵的专业知识,这在与实验人员交流结果时尤其重要。预测肽与MHC分子结合的几种方法都采用这种方法,例如[9-12],一项比较研究表明,如果数据量有限,则简单的统计方法生成矩阵的性能会比更复杂的人工神经网络更好[13]。
使用这样的线性模型隐式地假设可以认为序列中每个位置的残基对测量值的影响都是独立的和相加的。 为了成功应用SMM方法,这必须是一个合理的第一近似值,即使并入了对系数也是如此。 这是与一般学习算法(例如神经网络)的主要区别,该算法原则上可以对序列和测量之间的任何功能关系进行建模。
3.2 定量数据
作为SMM方法的输入和预测输出的实验测量是定量的,而不是二进制的。 例如,在肽与MHC分子结合的情况下,使用量化结合亲和力的IC 50值,而不是分类为结合和非结合肽。
如果定量数据的不同表示形式(例如IC50或log(IC50)值)是可能的,则应选择这样一种表示形式,即ymeas值近似遵循正态分布。 否则,SMM预测是独立贡献的总和,因此自身大致呈正态分布,将无法很好地拟合实验数据。 在绑定亲和力的情况下,这意味着应该使用log(IC50)值,因为IC50值本身通常是对数正态分布的。
3.3 嘈杂的数据
实验测量结果不可避免地会包含噪声。在建立将测量值精确化的模型时,这将导致问题。因此,SMM方法包含一个正则化参数Λ(方程2),该参数对应于将w的“平滑”值的更简单解决方案优先于精确再现观察值的解决方案。在第一个SMM应用程序[1,2]中,Λ是一个标量,在这种情况下,此方法称为Ridge回归或零阶正则化。隐式选择一个标量值将假定输入序列中每个位置的信噪比水平大致相同。在当前版本中,也可以选择Λ作为依赖于序列位置的序列,有时也称为局部Ridge回归。如图3中的示例所示,这使Λ成为对角矩阵,其中将属于相同序列位置处的残基的所有Λi值设置为相同值。对于长度为N的序列,存在N个不同的Λi值。对于许多包含与MHC分子的肽结合数据的训练集,我们将使用位置特定矩阵Λi与标量Λ所获得的预测性能进行了比较。位置特定的正则化几乎总是会产生更好的预测。如果不同序列位置的影响变化很大,则差异特别大(数据未显示)。

3.4 有界数据 Bounded data
任何实验技术都会生成包含在有限范围内的测量值。例如,在许多生物学实验中,“零”测量值通常意味着实际值低于实验分辨率,而不是实际值等于0。类似地,超出预期灵敏度极限的非常大的值不再具有定量精度。灵敏度范围的上边界或下边界的这些数据点所传达的信息与定量值不同,但它们仍包含信息。在我们可获得MHC结合数据的情况下,大约20%的肽属于此类。
就我们所知,SMM方法是设计用来从此类边界值提取信息的唯一方法。这是通过表1中所示的新颖L2 范数完成的。例如,如果实验测量值对应于ymeas z的上边界,并且预测值大于z,则ymeas与ypred之间的距离是零。该规范具有有用的特性,即可以通过迭代过程将任何根据L2规范的分析解决方案转换为根据L2 规范的解决方案:首先,将所有包括边界值的测量都视为标准值,并且使用L2规范已找到。在第二步骤中,对于L2范数为零的每个ypred,ymeas值对,将ymeas值设置为其对应的ypred值。对于这些ymeas *值,距离L2(ypred,ymeas *)= L2(ypred,ymeas)。这些ymeas *值然后根据L2范数再次用于求解。重复此过程,直到ymeas *值不再改变为止,如图4所示。



3.5 随机肽库数据
如前所述,用作SMM方法输入的实验数据由与定量测量相关的相同长度的氨基酸或核酸序列组成。在设计实验时,要测试的序列的选择会例如通过在特定序列位置上过量或不足地表示残基,将偏差引入训练数据中。避免这种情况的一种方法是使用随机肽库,也称为位置扫描组合肽库,它们是相同长度的肽的混合物。在给定的文库中,所有肽在固定位置共有一个残基,在所有其他位置均具有氨基酸的随机混合物。例如,文库XAXXXXXX包含在位置2具有共同丙氨酸的8-mer肽。此类文库可通过将其测得的过程效率与完全随机化的文库XXXXXXXX的过程效率进行比较,直接测量其共同残基的影响。对于8-mer肽,160个文库实验足以表征每个残基在每个位置的影响。这种完整扫描的结果可以汇总在一个评分矩阵中。这种方法已经在许多不同的实验系统中成功使用[14-17]
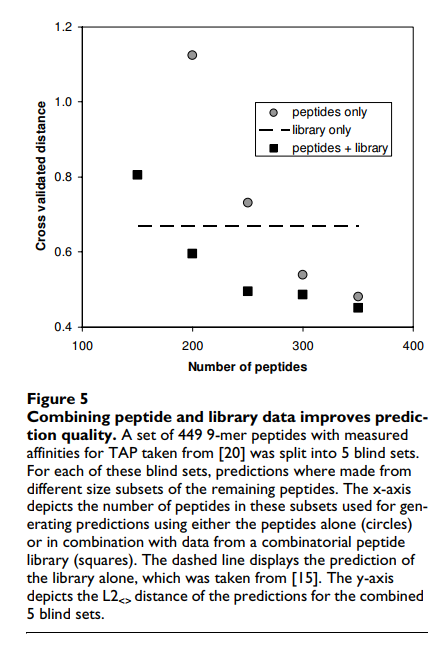
SMM方法的一个新颖特性是它可以合并来自这两个来源的数据。当给SMM算法提供来自单个肽段和随机矩阵的实验数据时,该库在评分矩阵matlib中汇总,它简单地从每个单个肽段测量值中减去matlib预测的值。然后将这些y’meas = ymeas-ypred,lib值用于生成第二个得分矩阵mat'。最终的SMM评分矩阵只是两者之和:matcombined = mat'+ matlib。图5将这种组合方法的性能与仅基于肽或文库实验的预测性能进行了比较。如果存在足够的肽段数据(大约与基质参数相同的数目),则组合的预测要比单独的库基质更好。在所有数据点上,组合的预测都比单独使用肽更好。重要的是,这种减去库预测的简单策略可以与任何预测方法结合使用,并且可能会产生相似的结果,因为它有效地增加了训练集的大小。为了可视化与图5中报告的距离相关的预测质量,图6描绘了对应于图5中具有最小距离的数据点的单个肽的预测和测量的结合亲和力的散点图。
3.6 引入对系数 Introducing pair coefficients
对系数可量化一对残基对测量值的贡献,该残差对偏离其各个贡献的总和。 如果对系数以与各个系数相同的二进制表示法引入,则等式(1)的形式保持不变。 图2给出了一个示例,如果考虑到两个这样的配对系数,如何将一组核酸序列转化为矩阵H。 注意,可能的配对系数的数量非常大。 对于三个核酸的序列,已经有(3 * 4)(2 * 4)/ 2 = 48对系数。 对于9-mer肽,存在(9 * 20)(8 * 20)/ 2 = 14400对系数。 据我们所知,只有SMM方法和加法[18]明确量化了对系数的影响。
由于大多数训练集仅包含数百个测量值,因此无法确定所有对系数的精确值。为了克服这个困难,SMM算法限制了要确定的配对系数的数量。首先,仅考虑存在足够训练数据的系数。根据经验,在训练集中必须存在5个在相同位置包含相同残基对的序列,才能考虑对对系数。在第二个滤波步骤中,仅保留训练集中信息相对合理的配对系数。在以前的SMM版本中,这通常由对系数的多次拟合确定,并丢弃观察到符号变化的系数。在当前版本中,使用了一种更快的方法。首先,为训练集计算得分矩阵,而无需任何成对系数。然后,针对每对系数,比较包含该系数的序列的预测值和测量值。仅当足够大的多数(> 60%)测量值高于或低于基于矩阵的预测时,对系数才会保留。其余对系数的确定完全类似于评分矩阵本身,但具有标量Λ值。
我们测试了将成对系数并入预测质量的效果,相比之下,对于许多包含肽与MHC结合数据的训练集,单独使用得分矩阵即可。对系数对大型训练集显示出一致的正贡献,其中训练量大于得分矩阵系数数量的1.5倍。然而,正如之前在[1]中所报道的那样,改进很小。如果评分矩阵的简单性比预测质量的小幅提高更有价值,则可以忽略对系数。
如果预计更高阶的序列交互作用(例如,由成对系数描述的交互作用)将对实验结果产生主要影响,则其他预测方法可能比SMM方法更适合。例如,通过选择与二进制向量不同的序列表示,可以对训练集中的信息进行概括,从而有效地降低输入参数的自由度[19]。即使输入数据有限,这也允许应用通用的高阶学习算法,例如人工神经网络。
四、结论
SMM方法可生成生物过程序列特异性的定量模型,进而可用于理解和预测这些过程。 相对于其他针对三种特定类型的实验数据的预测方法和工具,该方法先前已显示出很好的效果[1-3]。 但是,由于两个主要问题,很难概括不同方法之间的比较。 首先,经常无法获得在不同研究中使用的训练数据集,因此,在比较由不同方法生成的工具时,往往不清楚是由于一种更好的方法还是一种更好(更大)的训练数据集而带来的良好性能。 其次,从同一训练集生成工具可能很困难,因为使工具可用的出版物通常仅描述所用方法的基本原理。
为了克服第二个障碍,我们在这里介绍了一种实现SMM方法的计算机程序。致力于确保程序健壮,文档化,跨平台兼容并生成合理的输出而无需其他参数。同样,删除了以前使用的任何商业库,以允许代码的自由分发。这将允许任何感兴趣的用户以合理的努力来应用SMM方法,从而进行最重要的验证:应用于科学实践。
最后,我们相信本手稿中展示的两种策略与其他预测方法结合也将很有价值。首先,我们包含随机肽库收集的实验数据的策略可以直接转移到任何预测方法。当其他实验数据受到限制并且组合库中的数据可用时,这将始终对预测质量产生积极影响。其次,L2范数可以用作其他预测方法的误差函数。通过使它们能够处理实验边界值,这将增加需要定量输入的预测方法有效使用的训练数据量。
参考资料
- Peters B, Sette A. 2005. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics 6:132. 网址:https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-6-132
- Nielsen M, Lundegaard C, Lund O. 2007. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics. 8:238. (咱没有整理)
