【3】理解,预测和减轻翻译后物理化学修饰(包括聚集)对生物制药药品稳定性的影响
诸如单克隆抗体(mAb)的生物药物分子经历一系列复杂的处理步骤以获得最终产物。具体而言,基于蛋白质的生物治疗剂可以经历诸如生产,收获,纯化,重折叠,冻融,干燥,配制,褪色,雾化和运输的步骤以获得最终产品。这些加工步骤和因素,如高浓度,可变温度,极端pH值,变化的离子强度,搅拌,光照,剪切应力,气液界面和各种固液界面使药物分子受到许多应力和导致它们降解。分析化学的进步已经确定了蛋白质治疗中随时间发生的许多降解途径。这些途径不仅依赖于上述因子,而且还依赖于蛋白质序列结构,产生物理不稳定性或化学不稳定性,或两者兼而有之。物理降解途径包括展开,解离,变性,沉淀和聚集,而化学降解途径包括氧化,脱酰胺,天冬氨酸异构化和肽键水解。通常,蛋白质降解途径是协同的;也就是说,化学降解事件触发物理降解事件,例如当蛋白质中不稳定残基的氧化随后是蛋白质聚集时。这些降解不仅导致蛋白质药物产品异质性,而且当给予患者时可能导致免疫原性,减少靶标结合,改变药代动力学等等。
在本章中,讨论了当前用于预测降解的理解和计算工具,例如聚集,氧化和易脱酰胺位点。 因为在这些途径中,蛋白质聚集是最常见的降解途径之一,本章的大部分内容都致力于解决蛋白质聚集的计算工具。
一、蛋白质聚集 ( PROTEIN AGGREGATION )
单个蛋白质分子可以以几种不同的方式聚集在一起形成低聚物/聚合物,产生可溶性聚集体,亚可见的和可见的颗粒,纤维和沉淀物。已经在体内观察到蛋白质聚集(例如,在人类疾病中形成淀粉样蛋白,例如阿尔茨海默氏病,帕金森病和亨廷顿氏病以及在重组蛋白质的表达期间聚集)和体外(例如,在蛋白质产品的制造,储存和处理期间)。 由于体内和体外的蛋白质聚集都是一个重要的问题,因此对蛋白质聚集的主题进行了大量的实验和计算研究,并且他们广泛地确定了蛋白质聚集的一般机制:
- 原生聚集,即天然蛋白质的聚集;
- 非天然聚集,即构象改变的蛋白质的聚集;
- 化学修饰蛋白的聚集;
- 成核介导的聚集;
- 表面诱导的聚集。
从历史上看,由于其在神经退行性疾病中的重要性,蛋白质科学已经很好地研究了通过淀粉样蛋白样聚集途径聚集整合的蛋白质。在天然条件下(蛋白质最初折叠的条件)通过球状蛋白形成淀粉样蛋白需要蛋白质的全局或部分(或局部)去折叠。最近,通过天然途径的蛋白质聚集,其不需要蛋白质的部分或完全展开,也已经被研究用于生物药物
一般而言,蛋白质聚集的计算研究可分为两大类:(1)理解聚集机制的研究(2)预测蛋白质中潜在的聚集倾向区域(APR)的研究。
1.1 理解聚集机制的计算工具
已经使用分子模型技术(例如粗粒度模型(coarse-grained models)和原子模型(atomistic models))进行了大量计算研究,以阐明蛋白质聚集的机制和驱动蛋白质聚集的因素。 大多数这些研究都集中在已知通过淀粉样蛋白途径(amyloid pathway)聚集的小肽上,尽管最近的研究也涉及通过非淀粉样蛋白途径进行全长蛋白质聚集。
粗粒晶格模型(Coarse-grained lattice models)经常用于研究蛋白质聚集。在一项模拟研究中,使用简单的二维疏水极性(hydrophobic polar, HP)晶格模型来研究变性剂和蛋白质浓度对聚集的影响。这些模拟报道了聚集通过部分折叠的中间体的缔合开始,并且具有暴露于表面的疏水珠的天然HP蛋白易于聚集,尤其是当溶液条件有利于部分展开的构象时。
因此,增强分子内的疏水相互作用可通过稳定其天然状态来抑制聚集。在另一个模拟研究中,每种氨基酸的更详细的相互作用参数用于研究蛋白质聚集。 Cellmer等人使用重新归一化的Miyazawa-Jernigan势,这是基于知识的潜力,用于表示64-mer三维(3D)晶格蛋白的氨基酸与特定序列之间的相互作用。他们的模拟表明,较高的蛋白质浓度导致折叠景观的不稳定性和对错误折叠状态的偏好增加。这些错误折叠的状态伴随着链间相互作用的增加,这似乎弥补了链内相互作用的丧失。
原子模拟(Atomistic simulations )也被用于研究蛋白质聚集。 对AGAAAAGA进行全原子分子动力学(MD)模拟,AGAAAAGA是一种来自叙利亚仓鼠朊蛋白(ShPrP)的高淀粉样蛋白短肽(残基113-120)及其类似物AAAAAAAA(A8),显示β-片段低聚物含有6个到8 strands是稳定的。 另一项使用中间分辨率模型进行的MD研究表明,聚丙氨酸肽可以自发聚集形成淀粉样蛋白。 这些原子模拟也被用于探索序列背景对接种寡聚体大小和淀粉样蛋白形成短肽聚集构象的影响。 例如,原子模拟以及高温MD模拟正确地再现了Aβ16-22(KLVFFAE)的反平行β-折叠方向,如固态核磁共振( nuclear magnetic resonance,NMR )实验中所见。
还进行了原子模拟(Atomistic simulations)以研究环境因素的影响,例如肽聚集的pH。 在明确的溶剂条件下,在低,中和中性pH下对Aβ1-28和Aβ1-40肽的MD模拟显示两种肽在其等电点附近变得高度灵活。 由于疏水性尾部,Aβ1-40显示出比Aβ1-28更大的灵活性。 作者得出结论,由疏水性和电荷效应决定的构象灵活性是聚集倾向的主要机制决定因素。 已经进行了另一组原子模拟以阐明多肽主链和氨基酸侧链对聚集体的起始和传播的相对贡献。 一些研究已经证明,尽管多肽骨架驱动淀粉样变性,但特定的侧链序列可以影响聚集的动力学和热力学。
1.2 用于预测聚集区域的计算工具
大量的实验和计算研究表明,蛋白质上的一些(基于序列或结构的)特定区域驱动蛋白质聚集。这些区域称为聚集倾向区域(aggregation-prone regions, APR),往往具有独特的特征。电荷,疏水性,芳香性和二级结构偏好,可用于从蛋白质的其余部分描绘它们。生物信息学和现象学模型都已开发出来,使用训练数据集识别APR的独特特征,并利用这些知识预测蛋白质中未知的APR。这些计算方法中的大多数仅使用蛋白质序列作为输入来鉴定能够形成淀粉样蛋白样纤维的5至9个残基的短APR。基于模式识别,3D蛋白质结构和分子模拟的其他方法也正在出现。表3.1中提供了用于鉴定蛋白质中APR的可用计算机工具的概述,并且在下面我们更详细地讨论三种方法-TANGO,PAGE和SAP,因为这三种方法用于工程化生物治疗分子具有较低聚集的应用倾向已发表在文献中。
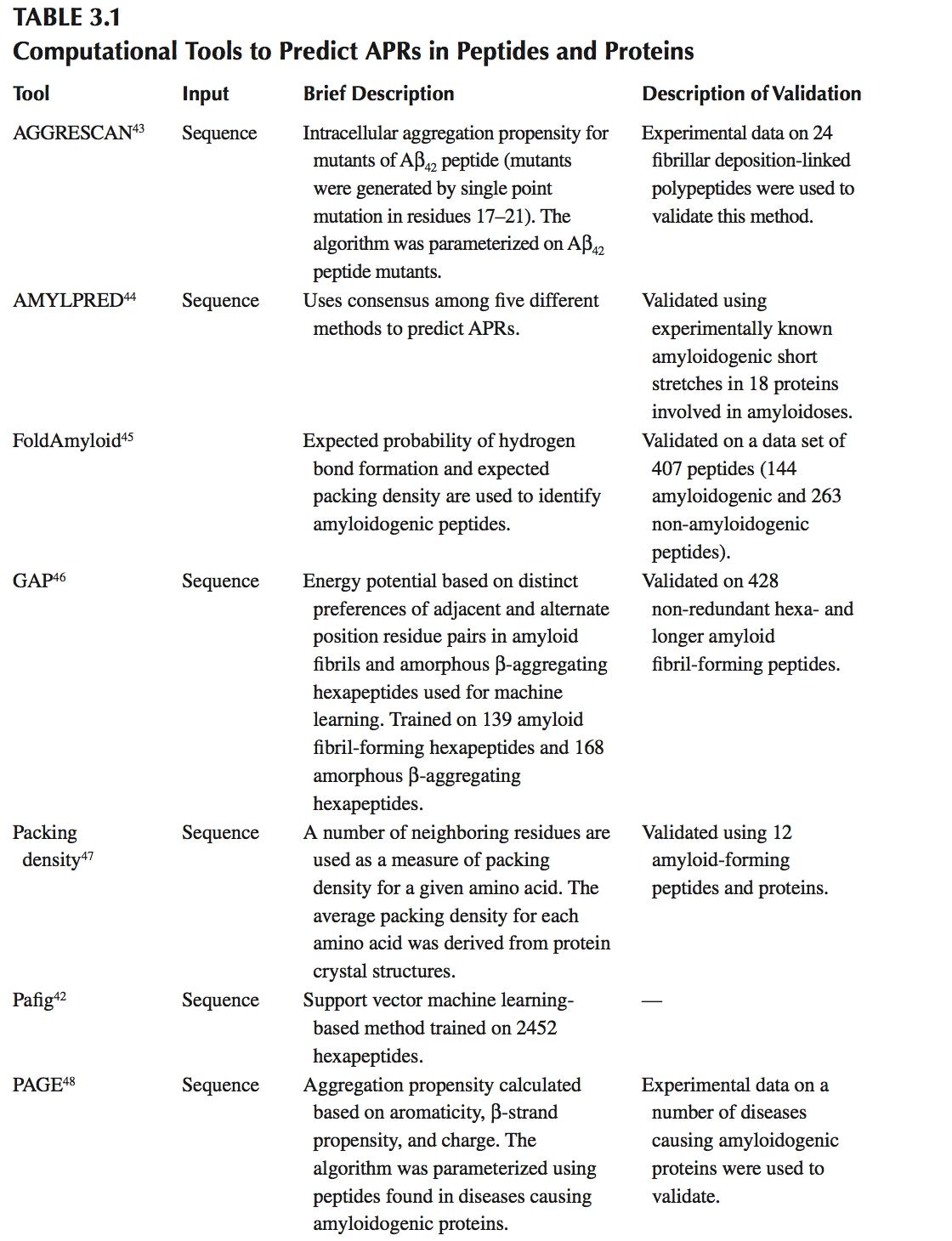
1.2.1 TANGO and PAGE
TANGO和PAGE都是基于序列的APR预测生物信息学工具。虽然这些工具已经针对淀粉样蛋白形成蛋白进行了广泛验证,但最近的一些研究也表明这些工具成功应用于生物治疗分子如抗体。 TANGO是一种基于统计力学的算法,它根据β-折叠形成原理计算聚合分数。它假设成核β-聚集区的核心区完全埋在聚集核中并满足它们的氢键电位。该方法还考虑竞争性构象状态,例如α-螺旋,β-链,转角,无规卷曲和β-聚集体。除蛋白质序列外,该方法还在计算聚集倾向得分时考虑环境因素,如pH,蛋白质浓度和离子强度。与TANGO一样,PAGE也是一种基于序列的工具,它通过沿着蛋白质序列滑动五到九个残基的小窗口来预测聚集倾向和绝对聚集率。通过PAGE计算的聚集分数基于芳香性,β-链倾向,电荷,溶解度以及给定窗口中每个残基的平均极性/非极性可及表面积。
1.2.2 SAP
与TANGO和PAGE不同,SAP(即,空间聚合倾向,Spatial Aggregation Propensity)是基于结构的APR预测工具。 SAP识别动态暴露蛋白质表面上的疏水斑块作为APR。 使用蛋白质3D结构作为输入,该方法鉴定疏水性贴剂,即暴露的疏水性残基簇。 由于该方法基于蛋白质结构,因此通过分子动力学模拟产生的结构集合也可用于SAP中以鉴定在正常蛋白质波动期间暴露的疏水性贴剂。 SAP鉴定的APR不是淀粉样蛋白形成序列模式; 事实上,这些APR由在氨基酸序列中可能或可能不是连续的残基组成。
1.3 计算策略优化稳定生物治疗
通过计算工具识别APR,可以用于设计聚集稳定的生物治疗分子。 这些计算方法可用于设计聚集稳定的蛋白质,或从蛋白质库中鉴定最稳定的蛋白质。计算工具还可用于设计用于减轻蛋白质聚集的辅助物。
1.3.1 通过蛋白质工程减少聚集
可以通过计算工具识别APR,然后破坏这些APR聚集,而生成稳定的生物治疗分子。 然而,进行突变以减少聚集的明显问题是突变可以影响蛋白质与其配偶体的结合亲和力和蛋白质生物物理性质如热稳定性。 例如,为增加抗体的聚集稳定性而进行的抗体的互补决定区(CDR)中的任何突变都可能潜在地影响抗体对其抗原的结合亲和力。
到目前为止,仅发表了一些关于鉴定生物治疗分子(如mAb)中APR的独立研究。除了这些已发表的研究外,还有一些证据表明Lonza,Accelrys,Molecular Operations Environment(MOE)和Schrödinger的商用计算工具也可以识别单克隆抗体中的APR。在一项研究中,基于序列的工具如TANGO和PAGE被用于鉴定mAb中的几种APR。这些APR中的许多APR与mAb的抗原结合区域一致,因此建议采用合理的基于结构的方法来破坏这些CDR环路中的潜在APR。在另一项研究中,SAP是一种基于结构的工具,用于鉴定mAb中的几种APR。图3.1和3.2分别显示TANGO / PAGE和SAP预测的全长人IgG1 mAb的APR(蛋白质)数据库[PDB] ID:1HZH60)。 TANGO / PAGE和SAP都预测了一些APR。例如,Fc区中由TANGO / PAGE预测的APR 302-VVSVLTVL-309与SAP预测的含有L309的APR重叠。可以进行L309向Lys残基的突变以破坏该APR,从而稳定mAb以防止聚集。事实上,IgG1mAb的L309K变体在加速聚集实验中显示出增强的抗聚集稳定性。
通过突变SAP预测的APR产生的其他变体也显示出增强的稳定性。然而,这些变体中的一些影响抗体与其配偶体的结合。例如,尽管变体I253K显示出非常低的聚集水平,但它显示出与蛋白A的差的结合,蛋白A用于纯化抗体。类似地,对于另一种IgG1抗体,虽然CDR中SAP预测残基的突变改善了这些变体的稳定性,但大多数变体失去了它们与抗原的结合。
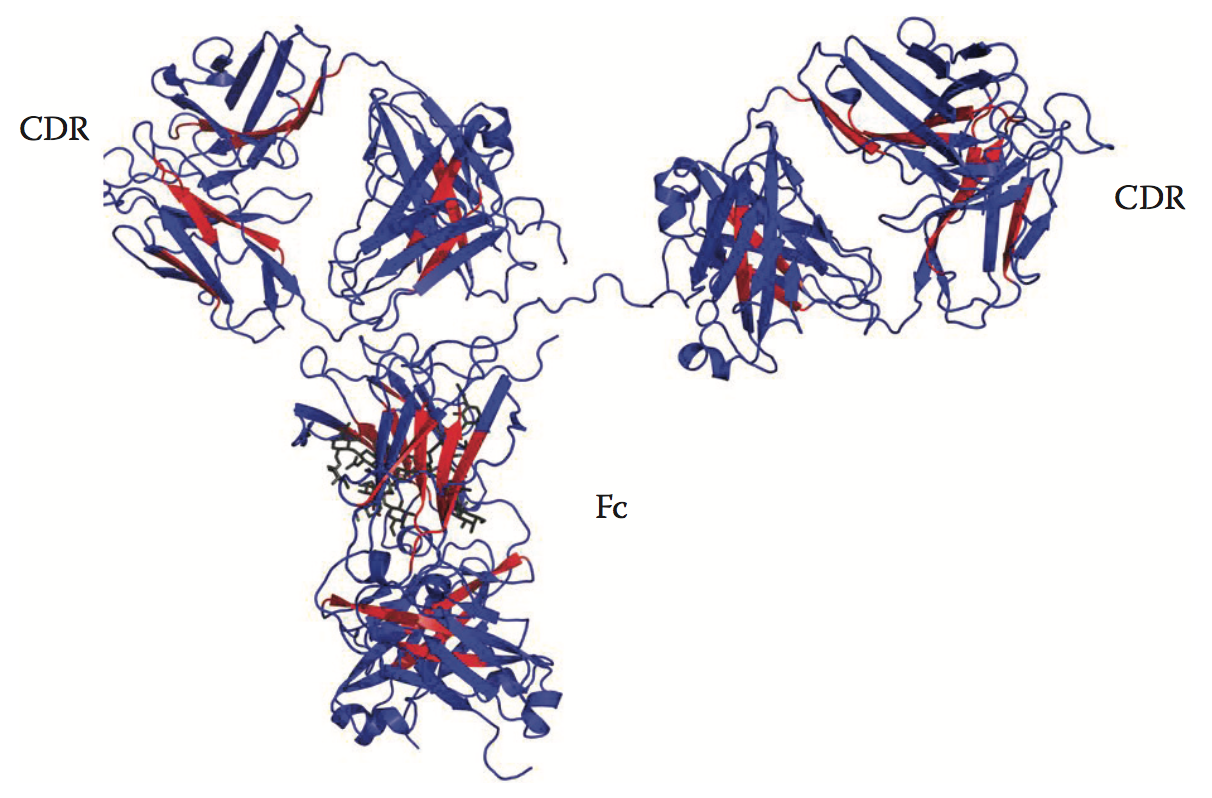
图3.1 The TANGO/PAGE-predicted APR (red) for antibody b12 (PDB ID: 1HZH60). (From X. Wang et al., mAbs 1(3):254–267, 2009.)
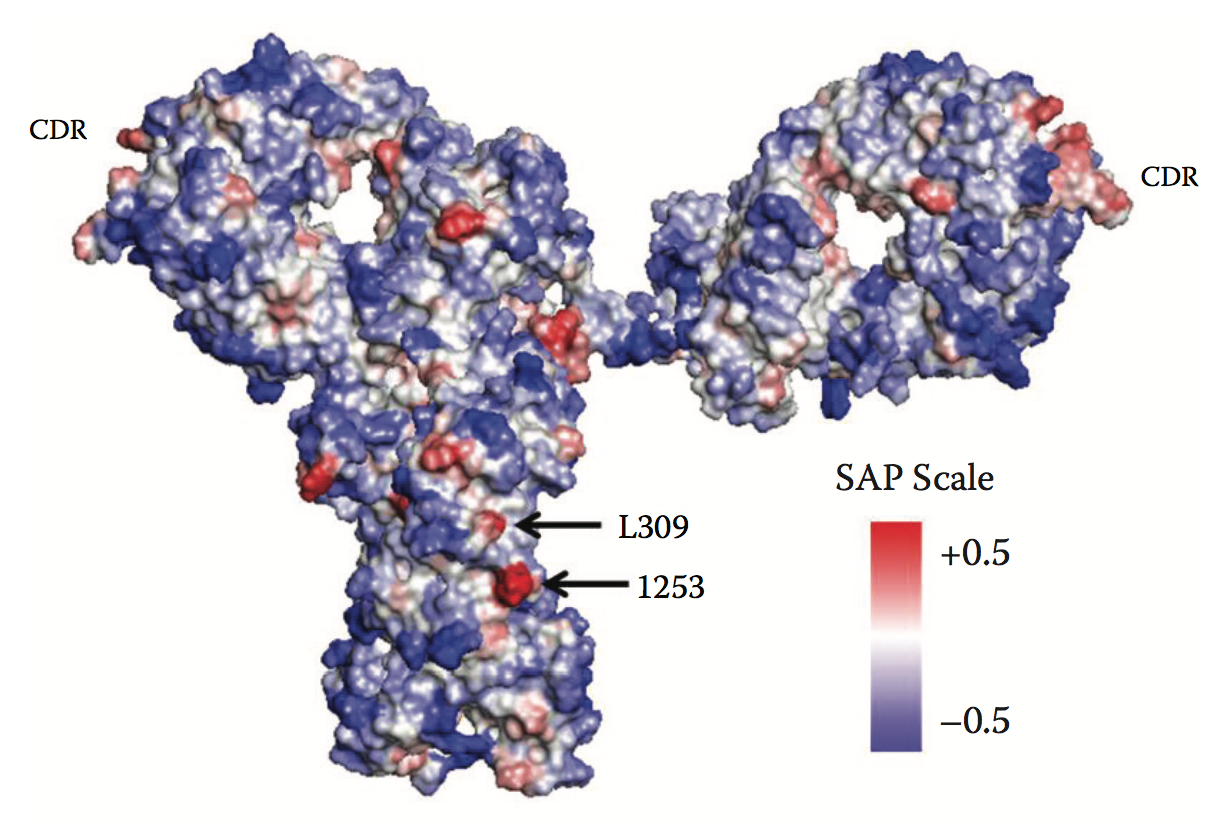
图3.2 The SAP-predicted APR (red) for antibody b12 (PDB ID: 1HZH60). (From N. Chennamsetty et al., Proc Natl Acad Sci USA 106(29):11937–11942, 2009.)
1.3.2 通过筛选稳定蛋白质进行聚集缓解
通过计算工具识别APR还可以用于根据蛋白质的聚集倾向对蛋白质进行分级,从而鉴定稳定的蛋白质。当APR存在于一个对蛋白质功能很重要的区域时,可以使用该策略而不是工程。例如,如前一部分所讨论的,对抗体CDR中存在的APR的工程化经常导致与抗原的结合丧失。在这种情况下,APR识别计算工具可用于执行从发现阶段出来的治疗性mAb的快速可开发性(或可制造性)筛选。在基于有限数量的mAb的已发表的研究中,SAP工具被扩展以根据它们的聚集倾向对这些抗体进行排名。这个扩展版本,称为可开发性指数(developability index,DI),结合暴露的疏水补丁(由SAP工具测量)和蛋白质净电荷的效果,根据其聚集倾向筛选蛋白质。这些计算工具可用于从发现阶段的mAb中鉴定稳定且不稳定的候选药物。稳定的候选人可以向前发展,而不稳定的候选人可以被丢弃或设计为稳定。
1.3.3 通过辅料设计减少聚集
赋形剂(excipients),例如糖,多元醇,氨基酸和聚合物,有助于稳定蛋白质溶液,特别是当蛋白质本身不能被修饰以减少聚集时。 当聚集倾向区域也参与抗原结合时发生这种情况(例如,抗体中的CDR可能负责聚集以及抗原结合)。 不同的蛋白质需要不同的赋形剂组合来稳定。 通常使用反复试验方法和经验推导的启发式方法选择这些赋形剂。 这既乏味又昂贵。 计算模型可以帮助合理选择赋形剂,并且可以机械地理解不同赋形剂如何在抑制聚集中起作用。
赋形剂的主要影响来自于它是否被蛋白质表面吸引或排斥。因此,如果蛋白质周围的局部区域中赋形剂的浓度不同于本体溶液中的浓度,则热力学性质的显着变化,会产生影响溶解度和构象稳定性的蛋白质。 使用优先相互作用系数(preferential interaction coef cient) Γ23来量化这种行为,Γ23是赋形剂对蛋白质表面的偏好的量度。 Γ23由以下表达式定义:
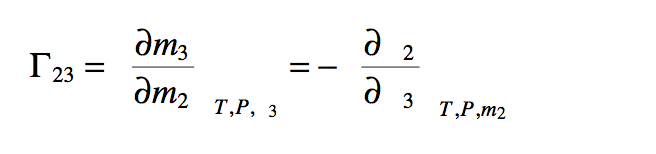
其中m,T,P和μ分别代表摩尔浓度,温度,压力和化学势,下标表示Scatchard符号中的溶液成分:水(下标1),蛋白质(下标2)或赋形剂(下标3)。由于局部结构域中共溶质浓度的增加,具有正Γ23的添加剂优先与蛋白质表面结合,并且如公式3.1所示,这种有利的相互作用降低了蛋白质的化学势。对于具有负Γ23的赋形剂则相反,其优先排除在蛋白质表面之外。优先结合或排除可以来自与蛋白质表面的非特异性相互作用(例如,体积排斥或表面自由能的扰动)或与蛋白质表面的特异性相互作用(例如,静电相互作用,氢键,阳离子-π与芳香族残基相互作用,疏水相互作用和疏溶作用)。
计算的全原子模型(Computational all-atom models)用于计算几种蛋白质和赋形剂组合的优先相互作用系数。 例如,优先相互作用系数用于解释精氨酸和谷氨酸的赋形剂混合物对蛋白质聚集的协同作用背后的分子机制。 在这里,计算模型显示协同效应与等摩尔混合物中蛋白质周围精氨酸和谷氨酸分子数量的相对增加有关,因为当两种赋形剂都是蛋白质表面上的赋形剂之间的额外氢键相互作用。 当下,蛋白质周围存在这些额外的分子导致增强的拥挤,这抑制了蛋白质的结合。 因此,基于优先相互作用系数的计算机模型可以给出赋形剂效果的估计以及对所涉及的分子机制的了解。
二、蛋白质化学修饰
批准或在临床试验中的大多数生物药物具有某种形式的翻译后修饰(post-translational modi cation, PTM),其可以深刻地影响与其治疗应用相关的蛋白质性质。 这些PTM包括氧化,脱酰胺,糖化,糖基化,二硫化物加扰等。 这些PTM可能在治疗性蛋白质的发展构成重大挑战和影响稳定性,效力,药代动力学和药效学参数以及安全性和免疫原性方面。 PTM位点的实验检测通常是劳动密集型的并且受酶促反应的限制。 在这里,我们讨论为基于蛋白质序列或结构鉴定潜在PTM和降解位点而开发的计算建模工具。
2.1 氧化 OXIDATION
氧化是治疗性蛋白质的主要问题,其可能对其安全性和功效产生不利影响。 氧化可以在许多残基处发生,包括甲硫氨酸,半胱氨酸,色氨酸,酪氨酸和组氨酸残基。 其中最容易残留的是甲硫氨酸,它通常会氧化成蛋氨酸亚砜(图3.3)。 蛋白质通常具有多个甲硫氨酸残基,每个残基可以氧化到不同程度。 评估最容易氧化的残留物的实验方法,如肽图谱和液相色谱 - 串联质谱(LC / MS-MS),既繁琐又昂贵。计算模型可以补充实验,有助于预测最更容易被氧化的残留物。
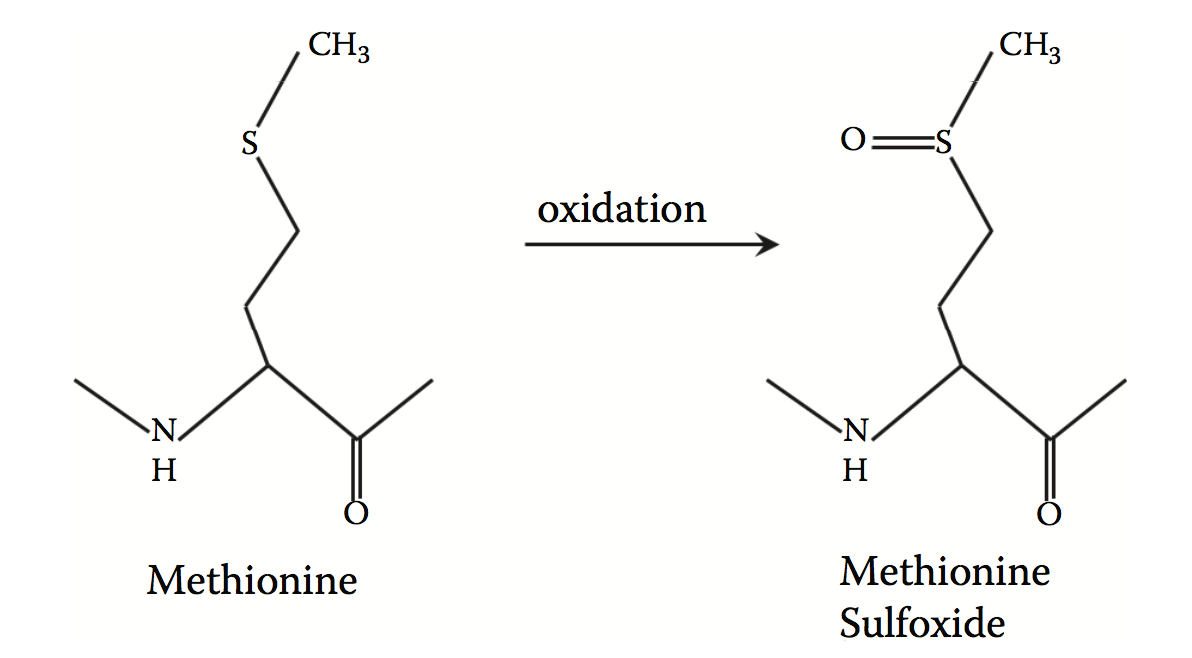
图3.3蛋氨酸氧化成蛋氨酸亚砜
甲硫氨酸氧化有多种来源,例如金属催化剂,过氧化物或暴露于光。氧化位点可根据氧化源而不同。金属催化氧化通常发生在蛋白质内的金属结合口袋附近(称为位点特异性氧化),而光致或过氧化物诱导的氧化可发生在蛋白质的任何地方(称为非位点特异性氧化)。在金属催化氧化的情况下,金属通常与Gly,Asp,His或Cys残基结合。一旦结合,在金属结合位点处产生活性氧物质(reactive oxygen species ,ROS),其可以氧化结合的配体(例如His或Cys)或其他靠近的易氧化残基(例如Met)。已经开发了多种计算模型来预测蛋白质中的金属离子结合位点。一旦用这些模型预测结合位点,结构建模可用于鉴定可能易于氧化的结合位点附近的Met,His或Cys残基。
过氧化物是另一种主要的氧化来源,尤其是蛋白质中的蛋氨酸残基。这些过氧化物可以来自配方或运输中使用的赋形剂,容器或管道。已显示过氧化物氧化受蛋白质结构特征的影响,例如蛋氨酸的溶剂暴露和蛋白质构象稳定性。因此,溶剂可及区域(solvent-acces-sible area,SAA)是预测最易氧化的蛋氨酸的最常用模型。该模型计算甲硫氨酸残基侧链的溶剂暴露表面积。通常,静态蛋白质构象(X射线或同源模型结构)用于计算SAA,因为其简单且易于使用。 Chu等人引入了另一种称为双壳水配位数的模型来预测最易氧化的蛋氨酸(图3.4a)。双壳模型计算了甲硫氨酸残基中硫原子的两个水配位壳(5.5Å)内的水分子平均数。 Chu等人表明,双壳模型与甲硫氨酸残基的实验氧化速率有很好的相关性(图3.4b)。
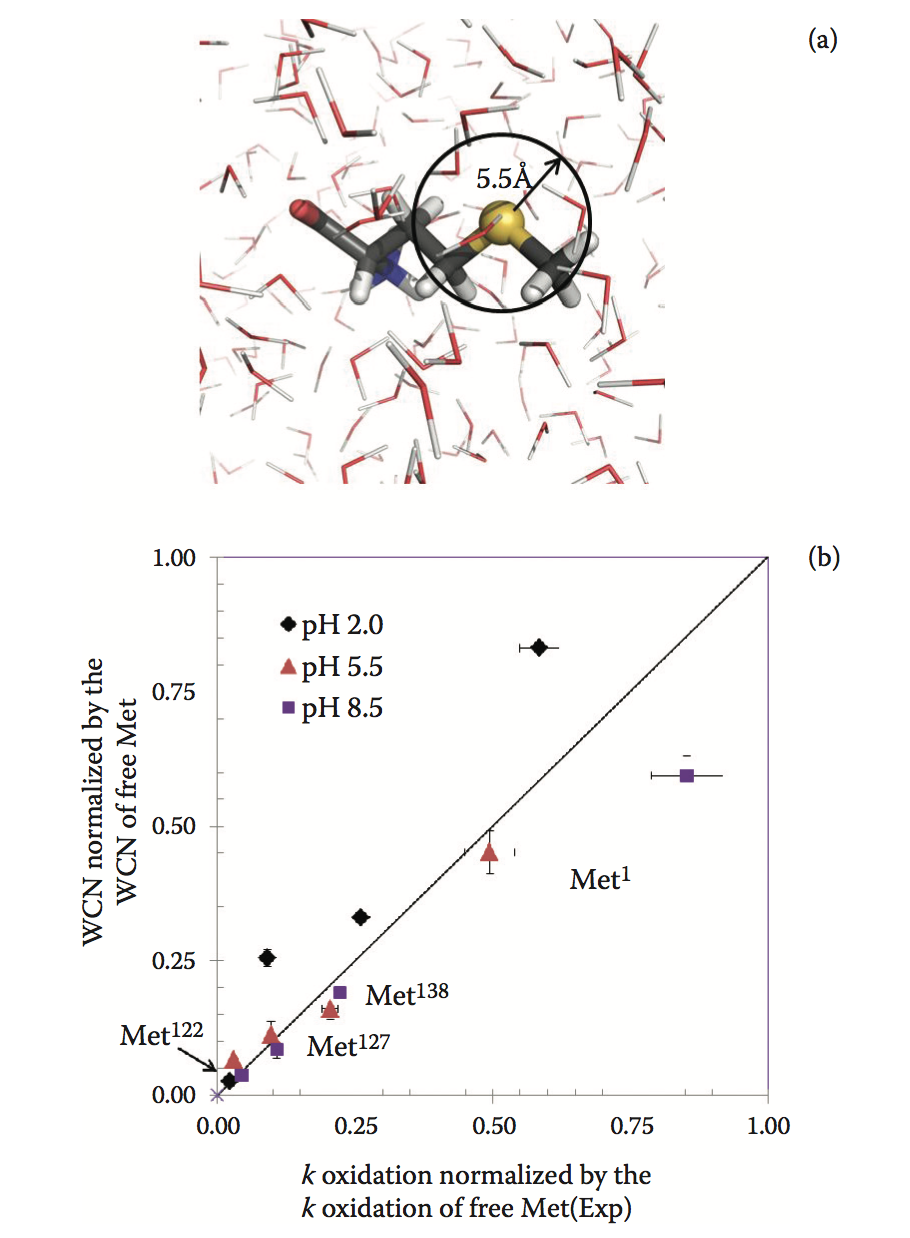
图3.4 (a)甲硫氨酸氨基酸的硫原子的双壳水(two-shell )配位数(2SWCN)的表示。 红线是水分子。 (b)甲硫氨酸硫原子的2SWCNs(截止半径5.5 Å )与不同pH值下G-CSF的甲硫氨酸残基氧化速率之间的相关性。 甲硫氨酸残基和蛋氨酸硫WCNs的氧化速率归一化为游离蛋氨酸氨基酸的值
最近的工作研究了双壳模型的准确性以及基于甲硫氨酸残基中的整个侧链或仅硫原子计算的几种SAA模型。这些模型目前通过七种模型蛋白和三种开发中治疗候选物进行评估。结果表明,基于模拟的双壳和SAA模型在预测由过氧化物引起的易氧化甲硫氨酸方面表现非常好。 此外,基于模拟的模型比基于静态结构的模型(例如X射线)更精确。 这些模拟模型不仅给出了相对氧化倾向的准确排序,而且与实验数据具有良好的半定量一致性。 因此,计算模型可用于识别最易氧化的蛋氨酸,这使研究人员能够通过直接诱变或配方优化来提高蛋白质的稳定性。
2.2 脱酰胺 DEAMIDATION
蛋白质药物中的天冬酰胺(Asn)残基可在制造和储存期间经历脱酰胺作用。 脱酰胺物种可导致蛋白质结构变化,聚集,药代动力学变化,活性丧失和潜在的免疫原性。 谷氨酰胺(Gln)残基也可以脱酰胺,但Gln脱酰胺速率比Asn慢100倍,因此通常不关心蛋白质药物。在中性或碱性pH条件下(pH> 7),脱酰胺作用 Asn通过称为琥珀酰亚胺的环状中间体进行。 然后该中间体水解形成天冬氨酸和异天冬氨酸的1:3比例的混合物(图3.5)。 在酸性条件下(pH <7),脱酰胺被酸催化,直接导致天冬氨酸。 已经使用从头算建模来表征涉及脱酰胺作用的反应网络的转变状态以及它们的pH依赖性
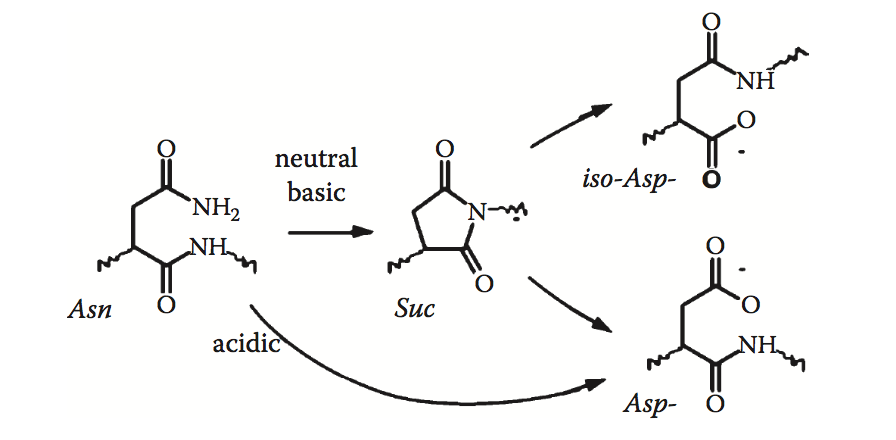
图3.5 天冬酰胺脱酰胺的中间体(琥珀酰亚胺)和最终(天冬氨酸,异天冬氨酸)产物。(Taken from B. Peters and B.L. Trout, Biochemistry 45(16):5384–5392, 2006.)
脱酰胺的速率随内在(一级,二级,三级结构等)和外在(pH,温度,缓冲液组成等)因素而变化。 确定一级序列对脱酰胺作用的影响的重要工作来自Robinson和Robinson。他们研究了Asn在五肽(Gly-Xxx-Asn-Yyy-Gly)中的脱酰胺率,并显示Asn脱酰胺的半衰期主要取决于 Asn之后的残留物。 Asn残基随后是Gly,His,Ser或Ala,具有脱酰胺作用最多的倾向。 这归因于残留物的小尺寸,例如Gly,Ser或Ala,其对分子内环化提供较小的空间位阻。
与肽相比,蛋白质中的脱酰胺率通常降低,因为灵活性降低。 此外,其他因素可影响蛋白质中的脱酰胺作用,例如与邻近残基的相互作用,包括氢键和环化的空间位阻。 蛋白质中的脱酰胺通常使用胰蛋白酶肽图谱进行实验评估,其可以给出脱酰胺程度的定量估计。 但是,这个程序非常繁琐。 因此,已经开发了计算建模工具来预测蛋白质结构的脱酰胺程度。 使用序列确定的五肽中Asn的脱酰胺速率以及几种蛋白质结构因子开发了一种这样的模型。 在该模型中,脱酰胺系数CD定义为
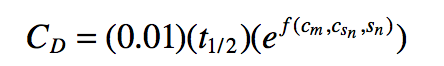
其中t1/2是五肽一级结构半衰期,Cm是结构比例因子,Csn是第n次结构观察的三维结构系数,Sn是观察结果,
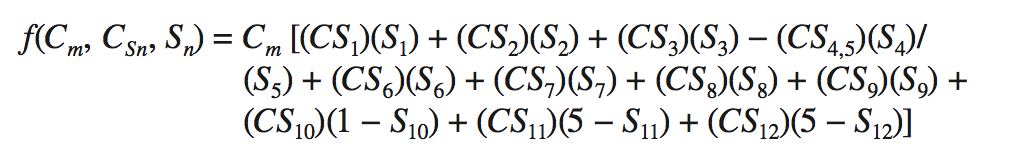
选择结构观察值Sn作为最有可能阻碍脱酰胺作用的结构,包括氢键,相对于α-螺旋和β-折叠区域的位置,以及缺乏灵活性的肽。该建模方法可靠地预测相对Asn脱酰胺率与23种蛋白质的实验数据一致。 Kosky等人使用多变量回归技术称为潜在结构投影(projection to latent structures ,PLS),以建立一组蛋白质物理化学特性与Robinson的氨基酸序列的脱酰胺半时间之间的数学关系(表3.1)。他们表明PLS模型具有降低倾向尺度,结合氢交换速率和灵活性参数,解释了脱酰胺半衰期中95%以上的序列依赖性变异。基于该模型,他们表明控制五肽内脱酰胺率的主要因素是酰胺质子酸度,亲水性,极化率和n + 1位氨基酸的大小。因此,计算模型可用于评估脱酰胺的倾向和获得机械洞察所涉及的蛋白质结构因素。
2.3 其他翻译后化学修改
糖基化( Glycosylation )是迄今为止最常见的与生物制药产品相关的翻译后修饰(post-translational modi cation,PTM),其影响蛋白质折叠,结合,运输,药代动力学和稳定化。糖基化是在蛋白质的特定位点上酶促添加糖,并且通常对蛋白质功能是必需的。最常见的糖基化是N-和O-连接的糖基化。通过将糖部分添加到天冬酰胺残基侧链上的氮原子来发生N-连接糖基化。通过将糖加入丝氨酸或苏氨酸的羟基发生O-连接的糖基化。然而,并非蛋白质中的所有Asn,Ser或Thr残基都被糖基化。因此,表征哪些位点是糖基化的是有用的,因为它可以影响蛋白质的稳定性和结合。对占据的糖基化位点的实验检测是昂贵且费力的过程。开发了许多计算模型和基于网络的服务器来预测糖基化位点,以及分析它们的结构。例如,开发了一个名为NGlycPred的模型,使用随机森林算法基于结构和残留模式信息预测N-连接的糖基化位点.IgG1抗体的结构模型显示其糖基化覆盖由以下组成的区域:几种疏水残基,如果暴露可能会导致聚集。此外,分子动力学模拟用于理解糖基化的影响。对IgG1和IgG2类抗体的模拟显示,糖截短或去除可导致蛋白质三级和四级结构的变形,可能影响蛋白质结合和聚集。因此,计算模型可以补充预测糖基化位点的实验,以及了解其对蛋白质结构和稳定性的影响。
糖化( Glycation )是另一种类似于糖基化的PTM,它还涉及向蛋白质中添加糖分子。然而,虽然糖基化是在蛋白质上的特定位点处的酶促添加,但糖化是非酶促的随意过程,其通常损害蛋白质功能。糖化发生在蛋白质上的伯胺位点,赖氨酸侧链或氨基N-末端。糖化导致形成席夫碱( Schiff base ),然后形成酮胺,两者都可以氧化和重排以形成晚期糖基化终产物( advanced glycation end products ,AGEs)。在正常衰老过程中,体内蛋白质自然发生糖化,并且AGEs在诸如糖尿病的疾病状态中更普遍。在治疗性蛋白质的情况下,糖化可以在体内循环期间或在细胞培养生产和制造过程中发生,其中它们暴露于糖。已经证明糖化在某些情况下会影响结合,而在其他情况下则不会影响结合,并且它有可能改变治疗性蛋白质的结构,功能和稳定性。因此,必须在治疗性蛋白质开发过程中表征糖化的位点和程度,以降低与产品质量和安全性相关的风险。结果表明糖化是依赖于位点的;某些含有赖氨酸残基的位点比其他位点更容易发生糖化。观察到许多因素影响糖化,例如pKa和赖氨酸的溶剂暴露,以及特定残基(包括天冬氨酸或其他赖氨酸)的接近程度。治疗性蛋白质的计算模型和分析显示,在某个位点是由于天冬氨酸残基的空间接近。另一项计算分析显示高度溶剂暴露的赖氨酸确实是高度糖化的。一种称为NetGlycate的模型是使用机器学习算法开发的,该算法以合理的准确度预测糖化位点。
二硫化物加扰( Disulfide scrambling )是当蛋白质内的Cys残基对之间的共价二硫键互换时发生的PTM。这种现象主要在IgG2和IgG4类治疗性抗体中观察到,但在IgG1类中相对较少。二硫化物加扰可以改变蛋白质构象和亚基缔合,并且可以导致产物异质性,可能导致药物功效丧失,解折叠,聚集和免疫原性。它显示导致治疗性IgG4s与内源性人IgG4交换Fab臂,并且它可能影响它们的药代动力学和药效学。计算模型可以帮助获得对蛋白质结构 - 功能关系的基本理解,包括理解蛋白质的共价二硫键结构变化背后的原因。使用原子分子动力学模拟分析硫原子的重复接近方法来自Cys残基对表示二硫键交换的可能性。例如,IgG2抗体的模拟显示铰链区动力学可以将Cys残基对置于大量空间上的近端位置,导致几种潜在的非规范二硫键结合方案,导致二硫化物加扰。因此,计算模型可以用于评估二硫化物扰乱的可能性以及由此产生的结构性后果。除了上面讨论的模型,还有几种计算模型和基于网络的服务器可用于预测其他PTM的位点,如磷酸化,乙酰化和甲基化(图3.6)。因此,本报告中描述的各种计算模型和工具可以补充实验和辅助 鉴定蛋白质序列和结构中潜在PTM的位点。

图3.6 可用作基于Web的服务器的计算模型,用于使用PTM预测蛋白质位点。 该图根据PTM的类型分离各种工具。 每种类型的PTM都提供最流行的预测变量。 (Taken from K.S. Kamath et al., J Proteomics 75(1):127–144, 2011.)
三、总结
在本章中,我们提供了用于识别蛋白质中各种物理化学降解位点的计算方法和工具的综合调查。由于它们与实验相比的速度和相对低的成本,这些用于识别易于降解的位点的预测工具可以证明在生物治疗发现和开发中非常有用。例如,可以在发现过程中尽早使用这些预测工具来设计易于降解的位点。与计算错误方法相比,由计算工具引导的蛋白质的合理工程化在降低易降解位点方面成功的可能性要高得多。当这些网站的工程设计不可行时,可以使用这些工具根据其降解liabilities对大量候选物进行排名。只有少数筛选出的degradation liabilities的候选物可以通过实验研究,以确定进一步发展的主要候选物。在生物制药工业中蛋白质发现的早期阶段解决蛋白质翻译后修饰的合理方法可以导致更多可开发或可制造的治疗候选物。在药物发现和开发的早期阶段解决这些修改有可能降低蛋白质药物带给患者的相关经济和时间成本。
参考资料
- 《Developability of Biotherapeutics》
