【9】评估生物制药候选物可开发性的最佳实践2(Best Practices in Assessment of Developability of Biopharmaceutical Candidates)
一、前言
生物制剂(Biologics)正在所有主要的制药和生物技术公司组合中获得动力,正在开发的产品范围广泛,包括抗体,生长因子,酶,多肽和毒素。生物制剂的成功开发不仅取决于药物改变生物途径的能力,从而产生治疗效果,还取决于药物开发者配制和制造药物的能力。不希望的生物物理和生物化学性质,例如差的溶解度和稳定性,聚集倾向和化学降解,可能影响功效并产生不需要的毒性反应,例如免疫原性,导致药物在后期开发和临床试验中失败。因此,在早期发现期间选择的具有次优生物物理和生物化学性质的分子转变为晚期开发,增加了磨损率(attrition rate)并且进行昂贵且耗时的制剂和开发活动。
由于这些原因,并且随着越来越多的生物制剂进入市场并且许多其他生物制剂在开发和临床试验中失败,生物药物开发近年来经历了范式转变。 在新模型中,更多基于知识的设计原则和实验筛选选择可开发分子在发现过程中尽早实施,而不是在配方和后期开发中修复“表现不佳”的线索。 显然,设计和早期选择适合进一步开发,配制和制造的候选药物可以降低后期失败风险,从而降低损耗率,降低开发成本并缩短生产药物的时间表。
The term developability has been coined to encompass the design principles and experimental assessment of the characteristics a molecule should meet to be fur- ther developed or manufactured, formulated, and stabilized in order to achieve the desired therapeutic effects.
可开发性被创造为包括设计原则和分子应该满足的特征的实验评估,以进一步开发或制造,配制和稳定以实现期望的治疗效果。 可开发性受许多因素的影响,包括分子的内在生物物理和生物化学性质,以及外在参数,如离子强度,pH和配方添加剂。
在本章中,我们描述了在生物发现和优化活动中执行的典型可开发性参数和测试。 应该注意的是,并非所有这里描述的分析方法都经常包含在可开发性评估中。 事实上,随着候选物选择的进行,进行越来越复杂的测试,对后期候选物进行更深入的审查。 我们还描述了最近开发的用于识别可开发性负债的预测方法。 这些方法在帮助设计更可显影的分子,和补充实验可开发性方法方面具有很大的潜力,以选择在后期开发中具有更高成功概率的分子。
二、可开发性评估的定义和药物发现过程
可开发性评估在发现和优化过程的几个阶段进行,包括一组实验和预测方法,旨在评估潜在药物的内在生物物理特性及其适应与现有制造平台相容的生产过程的能力。 尽管生物开发的可开发性评估尚未在整个行业中进行协调,但是对于一般最佳实践已经趋于一致,特别是对于治疗性抗体,其在当前发展中是生物制剂组合中非常重要的比例。
图9.1描述了典型的抗体药物开发过程。 它通过探索性研究开始(最重要的),重点是目标验证和assay开发。 在初始阶段之后,发现并优化潜在的leads,在将候选药物提交给监管机构批准研究性新药(IND)状态之前,将其发展为开发阶段的潜在候选药物。 早期发现(图中间),即未来候选药物的设计,主要集中在三个主要步骤:设计,筛选和选择。 在这些过程中,进行体外和体内测定以评估与靶标的结合,与直向同源物的交叉反应性,中和和与生物活性相关的其他参数。 此外,评估了四个可开发性参数:表达产率,化学稳定性,物理稳定性和溶解度。
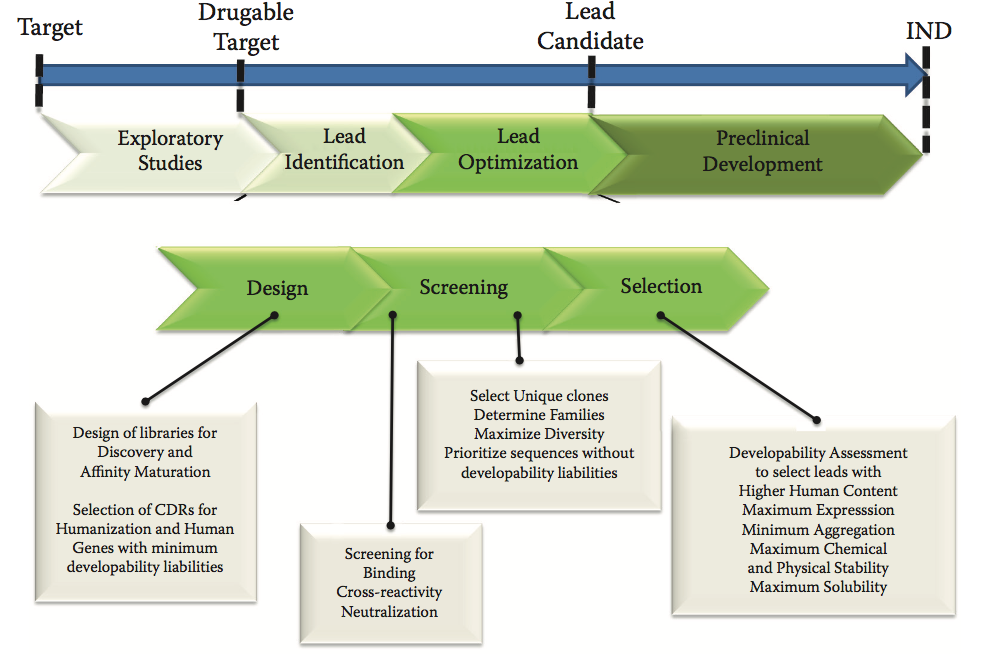
图9.1 药物发现和可开发性评估。
这些参数是相互关联的。 例如,化学不稳定性如氧化或剪切位点导致样品异质性,并最终影响物理稳定性或导致低溶解度或聚集。 不良的物理稳定性会暴露易于氧化或降解的侧链,最终在这些残基降解时导致聚集。 尽管如此,这些参数中的每一个都可以独立测量(表9.1),并且不符合成功标准的分子被优先考虑。 或者,如果鉴定出具有有希望的生物活性但在可开发性评估期间表现不佳的leads,则在优化阶段期间通过表的最后一栏中描述的方法将它们作为可开发性增强的目标。

为了在发现过程中尽早实验评估可开发性,必须重新设计工程分子的生物物理和生物化学特征的筛选,以使用少量蛋白质并且能够处理尽可能多的变体。 因此,期望可开发性评估方法适合于高通量筛选。 不幸的是,一些技术仍然需要大样品(有时超过100mg),因此不能用于早期可显影性筛选方法。
第4节中讨论的预测可开发性方法补充了在过程的每个步骤中应用的实验方法(图9.1中早期发现步骤下面的方框),甚至在发现活动之前,在设计阶段。例如,设计用于抗体发现的初始噬菌体展示文库以最大化在大肠杆菌中的表达。目前,用于发现的已经发展到在设计阶段包括预测方法,这些方法有助于选择可开发的人类基因,用作框架供体,以及选择可开发的互补决定区(CDR)。预测和实验方法的整合提供了对良好和不良行为的治疗候选者的详细评估,并加快了发现和优化过程。
三、评估生物学可开发性的实验方法
表9.2总结了在生物制剂的发现和优化活动中应用的实验方法。 如上所述,在候选开发的后期阶段执行增加复杂性或需要大量蛋白质的方法,而在早期候选筛选期间实施适合于高通量应用的更简单方法。 在以下部分中,我们描述了影响可开发性的机制,并讨论了评估它们的实验方法。

3.1 化学稳定性
生物制剂的化学稳定性已成为广泛研究的主题。 化学降解通过不同的机制发生,例如侧链降解或主链剪切。 侧链的降解影响多种氨基酸。 例如,氧化可以影响Met,Cys,His,Trp和Tyr; 水解影响Asp-Pro,Asp-Gly和Asp-Ser; 脱酰胺改变了Asn和Gln的化学结构以及Asn和Asp的异构化; 外消旋化影响His,Asp和Ser; β消除影响Cys,Ser,Thr,Phe和Lys。 此外,由于Cys残基的不正确配对和聚糖脱唾液酸化导致的二硫化物加扰影响了生物制剂的稳定性。
据报道,这些修饰中的一些会导致部分或完全丧失活性,增加聚集倾向,缩短产品的保质期,并增加样品的异质性。 对于抗体,除了参与抗原结合的残基侧链的降解(其可导致功效降低或丧失)之外,已经证明恒定区中的几个残基是潜在的降解位点。 这些残留物总结在图9.2中。 随着在不久的将来设计和开发更多的治疗性抗体,预计观察到的修饰列表将会增长。

图9.2 记录人IgG1中的序列倾向性。
3.1.1 脱酰胺和异构化(Deamidation and Isomerization)
Asn脱酰胺和Asp异构化是常见的降解过程,其涉及分子内反应,生成天冬氨酸和异天冬氨酸,通常以3:1的比例。 虽然有几个因素影响这些降解途径,但高(> 8)pH被认为是脱酰胺的常见原因。 已经对可溶性肽进行了几项关于脱酰胺速率的研究,尽管在全长蛋白质中,局部三级结构已显示出显着影响脱酰胺速率。 Gln脱酰胺遵循类似的机制,并且还报道了强烈影响三级结构并及稳定性。
Asn和Gln脱酰胺速率均取决于侧翼氨基酸的性质。 对于Asn,可溶性肽的研究表明,Asn残基的C末端残基在脱酰胺反应中起关键作用。 Asn和Asp,其次是Gly,His,Asp,Ser或Ala,被认为是潜在的脱酰胺位点,NG是最敏感的。 已经设计了用于鉴定脱酰胺的预测方法,并且报告表明预测可靠性为约95%。 已知Gln脱酰胺相对于Asn脱酰胺以较慢的速率进行。 与Asn类似,Gln的N-末端具有低分子量阻碍的带电残基已显示加速Gln降解速率
对于抗体,已经显示在CDR中发生的脱酰胺作用影响抗HER2抗体的效力。对于几种抗体同种型,还报道了重链和轻链恒定区域中的Asn脱酰胺,其中最活泼的Asn位于序列GFYPSDIAVEWESNGQPENNYK中。虽然人IgG1在恒定区含有15个Asn,但这些残基中只有4个对重链中的脱酰胺-N275,N344(QDWLN,WESN)和N390(QPEN)敏感,而N137(VCLLN)在轻链 - 证明NG和NN序列对脱酰胺的高灵敏度。还报道了相同的Asn残基在CH3中的IgG2N386(WTNN)和N423(NWERN)以及恒定轻链中的N156(ERQNGVLN)中进行脱酰胺作用。在小鼠IgG1中,还报道了重链中的N141(SAAQTN)和轻链中的N161(ERQN)的脱酰胺作用。有趣的是,在CDR中注射含有Asn的单克隆抗体后,在动物研究中也观察到体内脱酰胺作用。可变轻链上的Q13(EVQLVESGGGLVQPGR)和Q82(NSLYLQMNSLR),可变轻链上的Q19(APYTFGQGTK),轻链CL上的Q199和Q366(NQVSLTCLVK)、CH3中的Q366(NQVSLTCLVK)和Q422(SRWQQGN)已经报道了某种程度的Gln脱酰胺作用。
与Asn脱酰胺类似,DG序列中的Asp残基可经历异构化为异天冬氨酰残基。 已经报道了在CDR中发生的DG序列,以及IgG1铰链中的D221的Asp残基的异构化。 由于脱酰胺作用导致Asp和Glu侧链上的负电荷增加,脱酰胺作用的第一个结果是电荷异质性的增加,这可以通过分析阳离子交换色谱(CEX)色谱法检测。 然而,脱酰胺位点的精确鉴定只能通过胰蛋白酶肽上的质谱法(MS)来确定。 出于实际原因,可开发性评估包通常包括早期阶段的分析CEX或毛细管凝胶聚焦(cGE)以评估总电荷异质性,而对每种降解物质的准确鉴定留待后期开发。
3.1.2 氧化 Oxidation
Met氧化对生物治疗药物开发的影响已被广泛记录。 代表性的例子是α-1-抗胰蛋白酶(a1AT),是丝氨酸蛋白酶抑制蛋白家族的成员和用于治疗遗传性肺气肿的丝氨酸蛋白酶抑制剂。 已知a1AT由于活性位点中关键Met的氧化而在低pH下经历完全失活。 对于抗体,显示Met氧化导致Fc的结构变化,其影响抗体的稳定性,如CH2的Tm降低。如所预期的,氧化样品显示出增加的聚集速率。
人IgG1在Fc中含有两个保守的Met残基,在CH2中含有M252,在CH3中含有M428,在一些同种异型中存在另外的M358。 由于这些残基位于蛋白A和蛋白G结合位点(CH2-CH3界面周围),它们的氧化已经显示出改变了对蛋白A和蛋白G的亲和力。此外,Met氧化已经显示出影响与neonatal Fc受体和Fc-γ受体相互作用。 在IgG2和IgG3中,除了M252和M428之外,具有较低表面暴露程度的两个额外的Met残基M358和M397以较低的速率氧化。 在IgG1和IgG2中,证明Fc中的Met氧化改变了与Fc-γ受体的结合,抗体血清半衰期显着降低。
Met氧化通过形成Met sulfoxide 而进行,所述Met sulfoxide 可以不可逆地进一步氧化成sulfone。 几种因素可以诱导Met氧化,包括暴露于金属离子(如从生物反应器的不锈钢容器中舔铁),轻质,过氧化物杂质或其他自由基源。 虽然这可能在储存和处理期间发生,但是在发酵期间似乎发生Met氧化的高风险,其中条件培养基中的铁浓度可以相对较高。
通过检查氨基酸序列可以非常早地鉴定结合位点中的Met残基并通过序列工程消除。 如果Met残基对于结合是关键的并且因此不能突变为另一种氨基酸,则可以通过使用抗氧化剂或螯合剂来防止氧化降解。 例如,已经研究了自由基清除剂如游离Met,硫代硫酸钠,过氧化氢酶和铂以稳定HER2抗体。 还使用了其他螯合剂,如EDTA,二亚乙基三胺五乙酸(DTPA)和去铁胺甲醇盐。
Trp代表另一种易于氧化的氨基酸。 当暴露于UV光时,Trp的吲哚环可以氧化,增加样品的不均匀性。 这种化学修饰与治疗性抗体的商业样品中的黄化有关。 由于Trp残基的降解不影响蛋白质的电荷,因此通常使用疏水相互作用色谱(HIC)或MS进行Trp降解副产物的检测。 重组抗体中Trp氧化的大多数报道涉及CDR中的Trp。
如果暴露CDR中的Trp残基,也可以影响分子的溶解度,导致非特异性相互作用。 事实上,CDR中存在Trp残基已显示产生多反应性抗体。 当Trp和Tyr残基在长CDR中组合时尤其如此。 人IgG1在框架区中含有几个保守的Trp残基:Fc中有8个,CH1中有两个,Fv中有两个,它们构成恒定和可变结构域的核心。 这些残残基不是溶剂暴露的,通常不会影响溶解度。
His残基也可以经历金属催化氧化成2-氧代 - 组氨酸,尽管His的降解在文献中受到较少关注。最近,已经报道了His220在IgG1铰链中的氧化和His-His交联。
3.1.3 N-和C-末端修饰
已知许多N-和C-末端修饰发生在蛋白质中。 在抗体中,N-末端残基通常是Gln或Glu,其通常环化成焦谷氨酸。 抗体中最常见的序列修饰之一是C末端Lys的加工。 经常观察到这种修饰,因此抗体工程组中通常的做法是去除C-末端Lys。
由于在细胞培养和样品处理期间发生的信号肽的错误处理或其他蛋白水解切割,可能发生抗体N-末端的其他修饰。 使用自动Edman N末端测序可以容易地检测N-末端的修饰,而C-末端修饰更难以表征并且需要基于MS的肽作图。 或者,由于大多数这些修饰改变了蛋白质的总电荷和大小,因此可以通过分析CEX,等电聚焦(IEF)或毛细管等电聚焦(cIEF)检测它们。
3.2 物理稳定性
不可逆的去折叠可以改变蛋白质的功能活性并导致聚集。 在晚期候选选择期间,通常在不同的温度,pH,机械应力和不同的制剂缓冲液条件下测试生物治疗候选物的稳定性。 这些研究可以为下游加工提供信息,并帮助制剂组选择在优选配方平台中表现良好的候选药物。
生物治疗剂在低pH下的稳定性对于抗体尤其重要,因为它们在用蛋白A亲和层析纯化期间暴露于低pH,并且在病毒灭活步骤期间。 已经广泛研究了低pH对抗体稳定性的影响。 通过将样品在所需pH下孵育几小时,然后进行一系列分析,包括分析性尺寸排阻色谱(SEC),聚丙烯酰胺凝胶电泳(PAGE)和活性分析,以检测聚集和蛋白质活性保留的可能增加。已知暴露于低pH会使抗体不稳定,导致聚集和错误折叠
进行另外的稳定性研究以评估候选药物,对储存和加工这种冷冻和解冻或低温冻干的抗性。 这些实验通常包括使样品经历几个循环的冷冻和解冻或冻干和再溶解,并分析样品的聚集物含量和生物活性。
最后,一些可开发性评估活动可能涉及测试候选药物对机械应力的敏感性。 蛋白质倾向于在空气 - 水界面聚集,在搅拌下可能发生起泡。 这些研究通过机械旋转或摇动蛋白质样品,并在不同时间间隔测量聚集体的形成来进行
3.2.1 热稳定性
通常估计生物治疗剂的结构稳定性,测量蛋白质(Tm)在不同制剂条件下的温度依赖性转变点。 这些实验通常使用差示扫描量热法(DSC)或差示扫描量热法(DSF)进行,这更适合于高通量设置。 在基本假设下,蛋白质的热稳定性被认为是结构稳定性的预测因子,在较高温度下表现出转变的蛋白质在生理温度和储存条件下也将更稳定。
抗体在~70°C和~80°C左右分别显示出对应于CH2和CH3的两个特征性热转变。尽管Fab结构域熔融带偶尔可能与CH2和CH3的条带重叠,但它通常表现为第三个band的 Tm随每种抗体而变化。 在可开发性活动中,通常施加60℃的最小Tm阈值作为最低热转变的通过标准。 不使用峰值最高温度(Tm),1%蛋白质展开的温度(T1%)可以提供更准确的稳定性估计。
3.2.2 长期稳定性
长期稳定性是药物在不同储存条件下在相对长的时间(数月)内保持在溶液中的能力。 因此,在不同温度下测试药物的变体,通常为2℃-8℃,25℃和37℃数周。 在不同时间点收集样品,并使用分析型SEC和十二烷基硫酸钠聚丙烯酰胺凝胶电泳(SDS-PAGE)分析聚集体的形成。 还可以进行功能测定以确保蛋白质活性的保留。 这些研究是时间和样本消耗,因此通常在候选物选择的后期进行。
3.3 溶解度
由于对生物药物的酶促降解的敏感性,使得口服递送成问题,绝大多数生物制剂目前通过皮下/肌内注射或通过静脉内输注施用。 因此,注射体积的限制需要高浓度的蛋白质。 例如,虽然肌内注射的典型体积约为5ml,但皮下注射只能在注射体积低于1.5ml的情况下进行。 因此,需要配制浓度高于100mg / ml的抗体并不罕见,尽管只有少数商业治疗性抗体已经在如此高的浓度下成功配制。 虽然在极少数情况下可以进行多次注射,但这是不可取的。 另一方面,静脉输注可以在较低浓度下容纳更大的体积。 然而,这种递送途径对于频繁给药不是优选的,因为它需要就诊。
从实验角度来看,通过将蛋白质配制在所需缓冲液中并使用超滤装置逐步浓缩样品来进行浓缩实验。 在该过程中,通过目视检查和通过可见光谱或专用装置,评估样品浊度来监测聚集水平和沉淀或相分离的出现。 理想地,还可以通过用于微米尺寸颗粒的光遮蔽(LO),和用于40至1000nm范围内的亚微米颗粒的纳米颗粒跟踪分析(NTA)来测量可溶颗粒的形成。
3.3.1 粘性 Viscosity
进行浓度研究以测试浓缩蛋白质药物的可行性,同时保持样品粘度低于某些阈值。 具有高粘度的样品是不希望的,因为它们可能难以用注射器操作,需要更大的针规,最终增加患者的不适感。 尽管经常使用50 cP的经验粘度作为皮下注射的上限,但建议将15 cP的较低粘度阈值作为生物制剂注射的最佳限度。
3.3.2 聚合 Aggregation
聚集是生物治疗药物开发中的主要问题之一,因为高分子量物质(HMMS)可以是免疫原性的。 聚集可以在生产过程的不同阶段发生,例如表达,纯化,配制,储存和施用。 在过去的十年中,大量的工作致力于阐明蛋白质聚集的机制。 然而,蛋白质聚集是一个复杂的过程,通过几种可能的机制进行。 它受不同因素的影响,例如pH,温度和机械应力,以及蛋白质折叠中的内在不稳定性。
聚合可以是多种多样的。 在最简单的形式中,HMA可以源自蛋白质表面上疏水性或带电斑块的存在,而更复杂的聚集形式可能涉及蛋白质的部分或完全解折叠。 通常,蛋白质聚集体可以是可溶的或不可溶的(通过演变成更大的颗粒)。 它们可以具有共价二硫键并因此出现在PAGE凝胶中,或具有非共价连接,因此在SDS-PAGE中不可检测。 如果蛋白质的生物活性结构域在聚集颗粒中没有完全变性,则聚集体可以保留活性。
评估生物治疗药物的聚合水平是一项监管要求,它是每项可开发性评估活动的一部分。 IV注射中对可见和亚可见颗粒施加了监管阈值。 尽管尚未确定对可溶性聚集体的确定的截止限,但通常将聚集体保持在5%以下。
评估蛋白质样品中聚集量的标准方法是分析型SEC。 然而,已知该方法易于产生误导性结果。 有些可能是由于色谱柱或仪器的其他组件保留了部分聚集体的可能性。 其他误导性结果可能是由于某些聚集体的可逆性质,这些聚集体溶解在缓冲液流中,导致低估样品中的实际聚集程度。 事实上,尽管SEC色谱柱中的固定相是化学中性的,但通常会观察到与蛋白质的一定程度的相互作用,树脂和流动相的化学性质都会影响结果的质量。
出于这个原因,强烈建议使用正交技术来验证SEC结果。 广泛认为分析超速离心可以更准确地估算聚集物含量。 然而,它需要专业人员和专用设备。 或者,比色杯中的动态光散射(dynamic light scattering,DLS)测量对高分子量物质的存在非常敏感。 DLS避免了低估SEC缓冲液流中可逆聚集体溶解所产生的聚集物含量的风险,并且适用于高通量应用。 此外,可以通过DLS检测由于其大尺寸而被SEC柱过滤掉的可溶性颗粒。
3.3.3 未配对的半胱氨酸
已经报道了一些关于生物治疗药物中未配对半胱氨酸的报道。 在可开发性评估活动中测试未配对的半胱氨酸并不常见。 然而,我们在此包括关于该主题的简短讨论,因为未配对或错配的Cys残基的存在可显着影响蛋白质折叠,功能和稳定性。
二硫键是维持蛋白质正确天然折叠的关键结构元件。 未配对的半胱氨酸是反应中心,其可导致共价聚集体的形成并降低蛋白质稳定性。 在某些情况下,可能会出现二硫键的扰乱并导致蛋白质错误折叠,例如干扰报告。 一些细胞因子需要半胱氨酸结来维持正确的折叠。
根据物种和同种型,抗体含有不同数量的链内和链间二硫键。 人IgG1具有12个链内二硫键和4个链间二硫键,2个在下铰链中,2个将HC连接到LC,这稳定了IgG结构。 在兔和鸡等物种中,也发现了保守的cys残基。 还报道了人和小鼠抗体的CDR中的一对Cys残基,其对于结合靶标是重要的。
尽管正确折叠的IgG1不应含有未配对的Cys残基,但在治疗性抗体中报道了游离硫醇的存在。 特别地,据报道,与抗原结合位点紧密相连的Fab中的二硫键C22-C96的未配对导致抗体效力降低。 因此,尽管已经报道了在CDR中含有Cys残基的治疗性抗体,但非保守性Cys,特别是在CDR中,由于它们的反应性和形成共价寡聚体的倾向而被认为是一种可能性。
在非抗体丙烯酰胺凝胶中通常可以检测到在抗体中形成聚集体的未配对的Cys残基,其中抗体片段以典型的梯形图案出现。 传统上,可以使用基于Ellman试剂的比色游离巯基测定,来检测未配对的半胱氨酸,有时也称为DTNB。 最近,市场上出现了更敏感的测量方法,提高了灵敏度。
有趣的是,仅在蛋白质折叠变性后才能检测到游离巯基的事实表明(1)游离半胱氨酸被埋在蛋白质中,(2)尽管缺少二硫键,抗体仍保持非共价组装。 这反过来表明天然条件下的游离巯基测定,可能无法检测到埋藏的未配对半胱氨酸的存在,并且在这些测定中可能需要样品解折叠。 尽管条件培养基或制剂缓冲液中痕量的还原性杂质可能会减少这些二硫键,但它主要影响铰链区中更敏感和溶剂暴露的链间桥。 因此,埋藏的未配对半胱氨酸的存在可归因于在内质网(ER)中蛋白质合成期间错配的二硫键。
在其他IgG同种型中也已鉴定出非经典二硫键结构。 IgG2抗体在铰链区含有两个额外的二硫键,并且已知会发生扰乱。 一个有趣的病例也由IgG4抗体代表,其在重链之间显示异常弱的二硫键,并且可以在体外和体内进行Fab臂交换。
3.4 糖基化 GLYCOSYLATION
糖基化在生物制药产品开发中起着至关重要的作用,因为它通常被认为是异质性的来源。 糖基化还可以影响治疗性抗体的稳定性,溶解度,半衰期和效应子功能。 重要的是,CDR中聚糖的存在可以削弱与靶标的结合,从而导致生物活性的丧失。 因此,阐明生物制药产品中聚糖的存在和性质是一项监管要求。
抗体在CH2结构域的N297处具有保守的N-糖基化位点,这对于抗体的效应子功能是关键的。 尽管可变区上N-聚糖的存在并不罕见,但大多数人种系序列在可变区上未被糖基化。 这些额外的糖基化位点,通过约20%序列中的体细胞突变引入。 有趣的是,在一些疾病中已观察到可变区中糖基化水平的增加。
原则上,非保守位点的其他聚糖可能不会降低蛋白质的生物物理特性或降低其效力。 事实上,聚糖可以改善蛋白质的溶解度,因为它们由大的亲水部分组成,并且在本质上它们已知掩盖疏水表面。 在抗体工程中,已成功尝试使用聚糖掩蔽疏水性贴剂,并改善蛋白质溶解度而不影响亲和力。 在非保守位点的糖基化也在商业生物治疗中观察到,例如西妥昔单抗,其在重链的可变区上携带额外的聚糖。
然而,通常认为在非保守位点中存在额外的聚糖是不合需要的,因为它增加了regulatory compliance所需的分析工作量。 抗体异质性源于聚糖可具有多种化学形式的事实。 例如,Fc聚糖主要是在第一个环上带有核心岩藻糖的双触角结构。 这些糖形式也可以具有二等分N-乙酰葡糖胺,并且一小部分具有末端唾液酸残基。 另外的变异性来自于半乳糖环上的截短,导致携带两个,一个或没有半乳糖残基的结构(称为G2,G1和G0)
尽管每种抗体糖型的详细表征,通常不是可开发性评估活动的一部分,但是当糖基化参与生物活性时可能需要一定程度的研究。例如,已知非岩藻糖基化抗体具有比其岩藻糖基化对应物高100倍的抗体依赖性细胞毒性(ADCC)。终末唾液酸化显示通过降低对Fc受体的亲和力而对ADCC产生负面影响,但也积极地增加静脉内IgG的抗炎活性。此外,末端唾液酸化影响蛋白质半衰期和体内清除。最后,分别显示缺乏末端半乳糖和高甘露糖可减少补体依赖性细胞毒性(CDC)并增加血清清除率。因此,当需要某些生物活性如ADCC和CDC时,需要对聚糖部分进行更彻底的分析,包括准确估计岩藻糖基化和唾液酸化的水平。
在生物治疗药物中也经常观察到Ser和Thr的O-糖基化。 例如,CTLA4-Ig Fc融合蛋白在铰链区的Ser129和Ser139处被糖基化。 与可从序列分析中容易预测的N-糖基化不同,尚未鉴定出O-糖基化的共有序列,并且预测方法仅限于粘蛋白样结构域的鉴定。
在治疗性蛋白质中经常观察到的稍微相关的聚糖修饰是赖氨酸和N-末端胺中的ε-氨基的糖基化。 已知这种修饰在哺乳动物细胞培养期间发生,其中葡萄糖的浓度特别高。 向伯胺中加入葡萄糖导致一个正电荷的损失,这可以通过cIEF和IEC检测到。
在可开发性评估中,可以使用cGE或HILIC对抗体糖型的异质性程度进行初步评估,同时更详细地进行聚糖触角结构的结构测定,糖型分布的定量评估以及糖基化的精确定位。 可以使用质谱方法在开发的后期评估位点。
3.5 静电特性和筛选色谱条件
许多方案通常包括在可开发性评估中,以研究候选药物与某些纯化方法的相容性。 不同的组织在纯化和配方方法上采用不同的制造平台,这些协议通常在发现和制造组之间进行协作设计。
这些研究通过筛选缓冲液,离子强度和pH的不同色谱条件,最终筛选不同的色谱树脂来进行。 评估净化效率与去除不需要的污染物有关,最常见的杂质是高分子量和低分子量物质,产品相关杂质(聚集体和修剪产品),宿主细胞蛋白质,内毒素,DNA,浸出 蛋白质A和培养基成分。 对于抗体,例如,蛋白A亲和层析之后通常是由正交方法组成的抛光步骤,例如阴离子交换层析(AEX)或流通模式的CEX。 因此,可开发性评估的一个共同步骤是,确定抗体是否适合在离子交换色谱(IEX)树脂与组织的制造平台兼容的流通模式与梯度模式下进行纯化
一些色谱方法的使用对蛋白质等电点施加了阈值。 这是因为蛋白质的静电特性(连同样品的pH和离子强度)将决定蛋白质对离子交换树脂的亲和力。 例如,可能需要高于8.5的pI以确保在中性pH下的溶解度并允许在流通模式中使用AEX抛光步骤。 因此,可开发性评估研究需要在等电点,电荷分布和滴定曲线方面表征蛋白质的静电特性。 这通常从理论上通过蛋白质序列进行,或者更准确地说,通过IEF或cIEF进行实验。
四、预测方法
预测方法的开发甚至可以在分子合成之前识别可开发性倾向,因此可以节省大量材料并且减少药物开发过程中的大量成本。 一些倾向可以通过简单检查氨基酸序列来预测,但更多时候,可开发性倾向的可靠评估需要三维(3D)结构。
典型的可开发性评估活动首先分析具有良好生化和生物学特征的序列,以确定潜在的可开发性倾向(图9.3)。 对于抗体,通过噬菌体展示活动或通过杂交瘤技术获得的Fv序列,通常在较大批次的抗体纯化之前可用。 因此,序列分析可以快速鉴定可变区中的倾向和潜在候选lead的抗原结合位点。
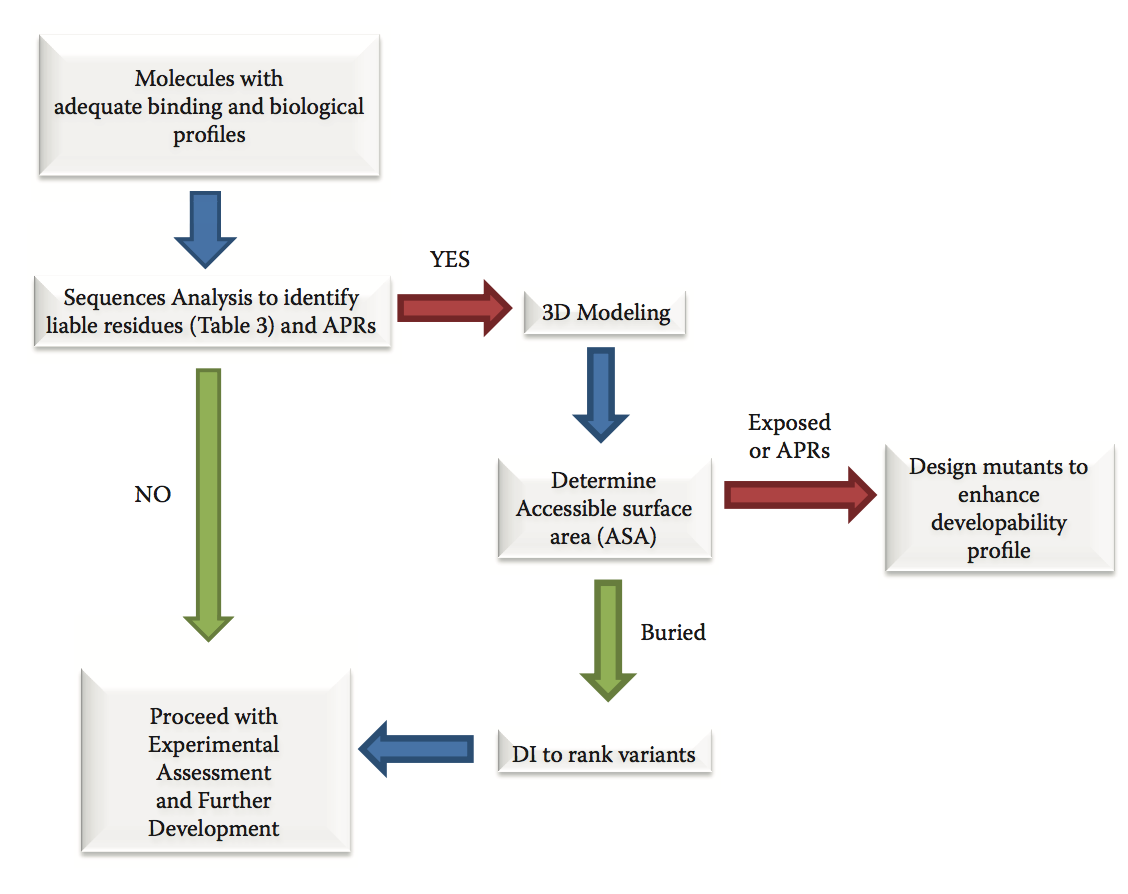
图9.3 在发现和优化过程中使用预测方法的工作流程。 红色箭头表示禁止使用。 绿色箭头表示去。 蓝色箭头表示过程的方向。
具有相似结合特征且没有明显倾向的序列,优先于具有可能损害进一步可开发性的残基的序列。 基于3D结构的更彻底的分析保留,用于具有有希望的结合特征但携带可疑残基的分子。 以下部分描述了在氨基序列中检测到的最常见的可开发性倾向,抗体3D建模的现有技术以及预测聚集的方法,这是生物开发的关键可开发性障碍之一,因为它可以导致免疫原性。
4.1 常见的序列倾向性 COMMON SEQUENCE LIABILITIES
典型的序列倾向已在前面的章节中讨论过,并在表9.3中进行了总结。 如果暴露于溶剂,表中列出的氨基酸可能经历脱酰胺,异构化,氧化和其他化学不稳定性。 一些残基,如Trp,可导致聚集,应进行进一步审查。 与难以预测的O-糖基化位点相比,可以容易地从序列预测N-糖基化位点。 此外,我们在“其他潜在倾向”下列出了从噬菌体展示活动中发现的抗体中发现的一些序列基序,这些基因已被发现与选择过程中经常使用的试剂交叉反应,如生物素,蛋白A或牛 血清白蛋白。
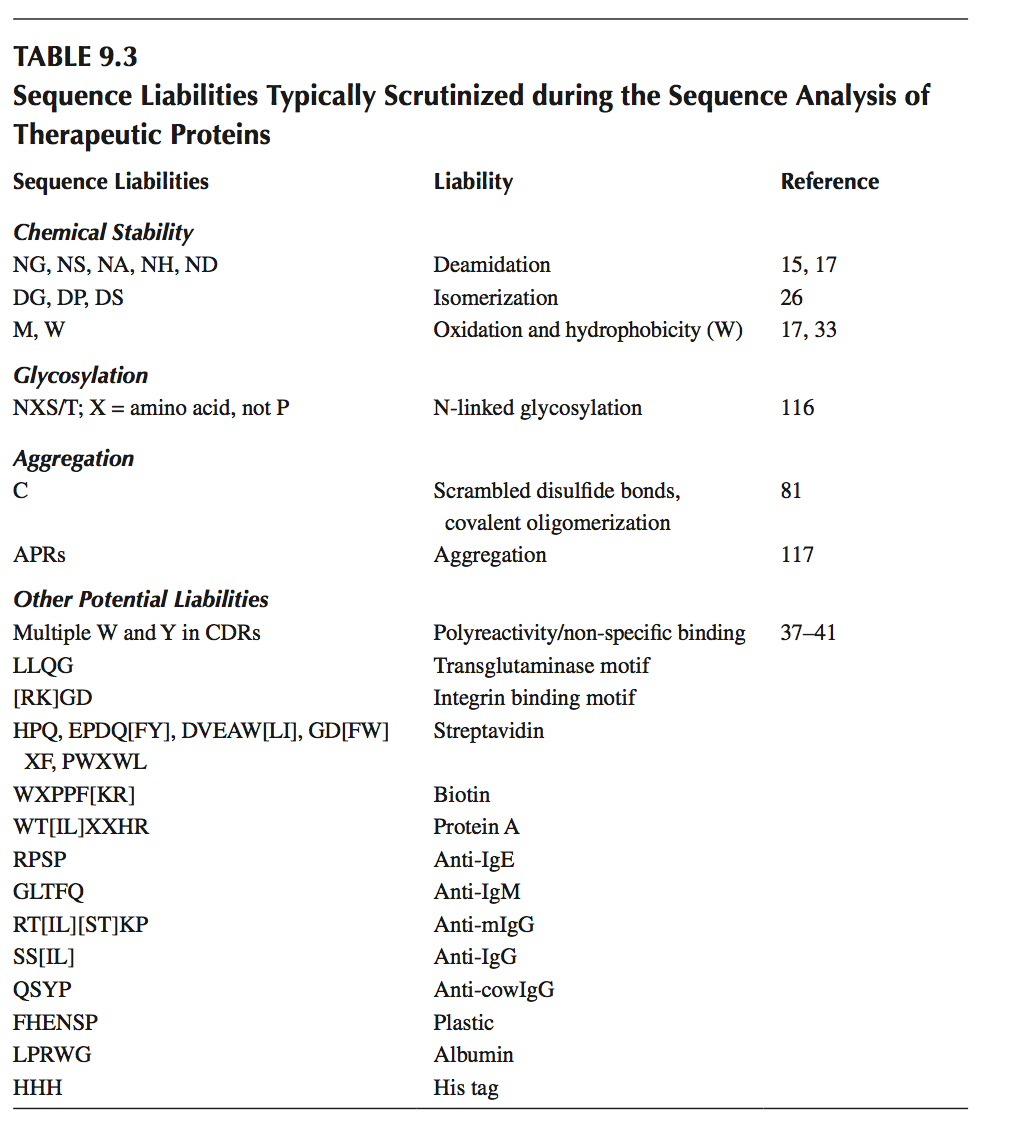
单独分析序列可能会过度预测化学不稳定性,因为可靠的残基可以埋在蛋白质中,从而防止降解。 另外,导致非特异性相互作用和聚集的疏水性贴剂可以通过序列中远离的残基形成,所述残基通过折叠(或解折叠)聚集在一起。 因此,3D结构的可用性对于加重可降解残留物的溶剂暴露和蛋白质聚集的倾向是至关重要的。
4.2 三维结构与建模
通过X射线晶体学确定蛋白质结构相对稳健,但是它是资源密集型的,并且在某些情况下,相对于计算建模方法,周转时间是不可预测的。 在没有实验结构的情况下,问题是3D模型如何与实验确定的高分辨率结构进行比较,因此基于模型的预测和设计的可靠性如何。
为了回答抗体3D建模的这个问题,其中一位作者最近比较了七种建模方法。 建模方法包括由Accerlys,Inc,Chemical Computing Group(CCG),Schrödinger,约翰霍普金斯大学的Jeff Gray实验室,Macromoltek,Astellas Pharma / Osaka University和免疫球蛋白结构预测(PIGS)开发的协议。 这些方法在许多方面都有所不同,包括自动化程度,用于模板选择的标准,能量函数的类型和采样算法。 尽管如此,所有评估的方法都产生了类似且可靠的Fv框架模型,并且除了少数例外情况外,还具有规范结构的CDR。
与第一次建模评估的比较也表明,在CDR-H3建模的过去3年中取得了进展,尽管它仍然是一个有改进机会的领域。 就序列,长度和构象而言,CDR-H3是迄今为止最可变的Fv区域,尽管已经描述了一些预测CDR-H3环的基础的规则,但没有可靠的规则来预测其构象。 已发现其严格基于序列模式的顶端区域(并且由于这些长肽链的内在动态特性可能不存在)。 因此,除了CDR-H3之外,Fv的3D模型是可靠的。
4.3 聚合和可开发性指数
聚集(Aggregation)作为生物制剂开发的主要关注点之一,已经成为许多研究的主题,并且近年来,已经开发了几种方法来识别易于聚集的区域。 一些方法,例如TANGO,Aggrescan,Amylprep和Zyggregator,是基于从短肽的淀粉样蛋白形成获得的知识开发的,称为易聚集区(ggregation-prone regions,APR)。 其他方法,例如空间聚集倾向(Spatial Aggregation Propensity,SAP),通过可及表面积和疏水性的组合预测易聚集的结构基序。
SAP还与估计分子电荷相结合,以开发所谓的可开发性指数(developability index,DI)。 DI估计长期物理稳定性,其被认为主要受分子基于溶剂暴露的疏水残基和分子的总净电荷聚集的趋势的影响。后一参数是在整个序列上计算的。通过SAP估计分子表面中疏水斑块的程度。由于DI提供了数字输出,因此该工具可用于在选择后对具有不同设计或Fv的蛋白质变体进行排序。由于分子的电荷随pH变化,DI也可用于估计不同pH条件下的聚集。应该注意的是,DI不考虑其他降解途径,例如氧化,水解,以及未配对的Cys残基和其他可能的残基的存在。因此,在设计或从候选药物库中选择lead时,需要考虑其他倾向。
五、结论和未来前景
可开发性评估过程是一个不断发展的领域,其中更多的实验测试被推向上游进入早期发现。最终,这将降低开发成本,并通过转换到符合某些质量标准且与某些纯化/配方平台兼容的仅后期分子,来避免后期失败。 目前,在开发的早期阶段开始的一些可开发性测试,受到某些技术不适合高通量多路复用应用的事实的阻碍。 在这种情况下,开发具有高通量模式的新型自动化仪器可以迅速改变可开发性评估实践。
由于速度和相对低的成本,预测方法有可能大大改善可开发性评估过程。 预测方法与高通量分析工具的整合将在早期提供对表现良好和不良行为的候选物的详细评估。 此类信息应加快发现,制定和制造过程。 随着开发中生物治疗药物的数量增加,并且引入了与传统单克隆抗体和Fc融合方式不同的新型支架,可开发性评估方案的设计也将改进并导致更强大的方法,统一标准和行业良好实践 。 目前,文献中有一些报告可用于报告可开发性策略,但关于该主题的大多数讨论都在快速迭代和发展中。
参考资料
- 《Developability of Biotherapeutics》
