【10】可开发性评估工作流程降低生物制药开发风险(Developability Assessment Workflows to De-Risk Biopharmaceutical Development)
一、简介:为什么要可开发? 风险是什么?
最近对开发新药的真实成本的估计表明,平均而言,公司似乎花费了40亿美元到110亿美元用于最终商业化的每种新治疗方法。 这是什么原因? 从根本上说,在药物开发中观察到极高的失败率。 大约90%的候选药物在临床开发过程中会失败 - 如果还包括临床前开发阶段,可能超过99%。 在这种情况下,人们越来越关注最大限度地提高新治疗候选人开发投资的回报率,避免任何可能的晚期和昂贵的失败。
在本章中,我们将讨论早期风险评估,特别是使用可开发性方法和计算方法,如何以经济有效的方式帮助降低开发过程中的风险。 我们定义了特定的风险领域以及它们如何影响广义的产品质量,包括产品功效和患者安全等基本方面。 介绍了围绕可开发性的新兴行业实践,包括生物制品应用的一些具体实例。 此外,我们建议工作流程示例说明在早期药物开发过程中如何在实践中引入可开发性策略,以降低风险,减少药物损耗,并最终提高生物制药供应链的稳健性。 我们还讨论了这些方法的实施如何也可以加速新治疗方法的临床应用。
1.1 为什么药物在发展过程中失败
药物开发的失败似乎是必然而不是例外。 然而,许多人在开发过程中药物失效背后的真正原因仍然是一个备受争议和难以捉摸的问题,这主要是由于缺乏关于该问题的详细和最新数据。 在某些情况下,药物候选药物中止背后的具体信息不会公开(通常在临床前开发期间)。 在其他情况下,不同元素的组合在特定候选药物的消亡中起作用,使得难以确定具体的促成因素。 来自Kola和Landis的出版物涵盖了1980年至2000年期间的药物开发以及自Arrowsmith和Miller以来发表的其他分析,这些出版物揭示了这一主题,表明了药物流失背后的不同原因。
显然疗效不足可能是临床失败背后的主要原因,但药物磨损的其他相关原因包括生物利用度和药理学缺点,安全性和毒理学问题,甚至稳定性和质量问题,后两者对生物制药尤其具有挑战性。此外,停产的战略和商业原因通常包含设计问题,商品成本,缺乏市场或正在开发的其他产品的竞争优势,甚至资金不足。尽管新药候选药物在晚期临床开发和注册过程中的失败主要与生物活性和功效不足,或药理学和剂量问题有关,但早期临床开发过程中的磨损从根本上涉及安全性问题(生物制药中的免疫原性反应)和药理学,后者在生物药物中较少见。如上所述,临床前药物损耗是一个非常复杂的调查领域,但根据我们的经验,与产品稳定性和生产率相关的制造和质量问题是观察到的常见问题。同样重要的是,质量问题是监管机构召回药品背后的主要原因之一,主要与不可接受的杂质水平,不稳定性或患者安全问题有关。
1.2 风险意识和可用性偏差: 为什么我们认为“它不会发生在我们身上”
有人可能会认为,我们的行业应该只是意识到最终落后于药物开发中观察到的极高失败率的许多风险。虽然制药行业已开发出高度复杂的流程来开发新药,但它对风险,发生可能性和决策过程的共同偏见并不免疫。其中最主要的是可用性偏差,通过直接经验或由于外部曝光,新闻,报告等相关突出,通过我们可获得的信息来表达我们对风险的看法。药物开发中不同职能的风险认知存在很大差异,特别是在评估不同药物失效原因的相对影响和突出程度方面。也许这是由于药物开发的复杂性,药物开发生命周期缺乏整体观点以及治疗发展的不同阶段之间现有的“divorce”所致。通常,第一手的失败知识可以真正帮助详细了解与之相关的后果和成本。下面我们列出了一些有助于增减风险及其潜在后果和影响的因素。
-
只做必要的事情。 开发新药是一个漫长而昂贵的过程,并且存在巨大的压力,需要快速,经济地进行。 但是,如果在发展的早期阶段没有适当考虑未来的风险,可能会造成巨大的后果,包括时间损失和额外成本。
-
抵制变革。 通常考虑采用新的方法或技术会导致行业担心,特别是在新药的开发和制造方面。 默认位置始终是使用经过试验和测试的方法,而不是冒着与监管机构发生潜在并发症的风险。 矛盾的是,事实证明风险缓解方法或更强大的制造平台的引入可被视为潜在采用者的高风险,他们倾向于认为当前发展实践的良好趋势是更安全和更简单的前进方式。
-
False economies。在开发中预先实施降低风险的努力往往被视为分散注意力,昂贵等等。这反过来又与我们刚刚提到的在后期发展阶段对质量失败的真实成本缺乏认识有关。例如,这些失败包括失败批次的成本,无法为期望的给药途径配制产品的成本,由于质量或安全问题而在临床开发期间取出产品时产生的累积成本,以及等等。 Juran对此非常了解并开发了一整套用于评估质量成本(CoQ)的语料库,其中包括一些人认为质量低劣的成本。不幸的是,这些未来事件并不总是容易发现,或者更糟糕的是,参与早期开发的科学家完全看不到,因此质量差(包括药物失效)的成本很少被纳入任何重大投资决策。
-
高度分散(silo)开发过程。 药物开发背后的典型层次结构,通常意味着完全缺乏对下游可能出现的问题的了解,但也迫使不同的产品和流程优化活动完全相互隔离。通常,期望下游的某个人会知道如何把事情做好。 因此,流程在每个silo内独立优化,而不是作为一个整体。 相反,为优化一个特定阶段而引入的变化可能对另一个阶段产生非常不利的后果(即,以牺牲患者顺应性或给药成本为代价,以杂质分布或制剂稳定性为代价进行滴度优化)。
-
过去的经历或“我们是不同的”信仰。 过去的经历往往会影响对风险的感知。 这在药物开发中尤其严重,其中候选药物可能需要8至15年的开发才能成为商业产品。 这意味着,科学家充其量只能有限或部分地接触某一特定产品的历史,以及与其发展相关的任何潜在问题。
1.3 每个的潜在风险:失败的代价
如上所述,目前分散的药物开发方法的悲剧性后果是,在新候选药物的发现和早期开发过程中,可能会遗漏对新疗法成功至关重要的设计要素。 流程优化和产品优化之间的这种根本性脱节产生了一个差距,可能导致后续的重大问题,这些问题通常只在开发的很晚才发现。 因此,可能会出现严重的延误,需要额外的投资,或者在更糟的情况下,会导致停止整个药物开发计划。 实际上,在开发和生产生物制品期间观察到的开发,返工,批次失败或偏差的延迟都是频繁且昂贵的问题,并且在许多情况下,最终可以追溯到候选产品或制造过程的不良设计。
此外,临床前和临床损耗的财务影响往往被忽视。 从生物制药开发的角度来看,在产品甚至被批准用于临床试验评估之前,已经为开发合格的制造过程做出了重大的财务承诺。 实际上,通常为原型(候选药物)开发完全商业定义的过程,在大多数情况下,它们在其开发周期的某些时候会失效。 这种投资显然存在风险,取决于各种临床前和临床开发阶段的成功。 此外,潜在的制造或安全问题也可能在以下几个方面产生重大的财务影响:
- 延长已经很长的开发时间(减少市场独占期)
- 需要在流程开发,重复工作或实施纠正措施方面进行额外投资
- 阻止项目停滞,而不能进入临床开发的后期阶段
- 导致临床试验期间程序失败,需要重复试验或因质量或安全问题阻止最终的商业批准
- 在流程重新设计和调整,重新制定甚至产品召回方面需要大量投资
这些问题可能值得数百万人失去机会或缺乏回报的投资。 在这种情况下,希望通过提出正确的问题来尽早选择或设计一个成功的候选物。
二、设计可开发性和质量(QBD)
2.1 可开发性(DEVELOPABILITY):另一个质量的词
在定义质量时,Juran在质量方面区分了小Q和大Q. 前者主要关注制造,制造过程和制成品,而后者,大Q,对质量有更全面的看法,涉及更广泛的流程,利益相关者和客户需求。 此外,大Q要求引入Juran所谓的质量三部曲:质量规划(质量设计的另一个名称),质量控制和质量改进。 在定义质量成本(CoQ)时,这种更广泛的质量定义(大Q)和强调质量计划(设计质量)也具有重要的内涵,特别是在了解制造失败批次之外的不合规的真实影响时。
ICH Q8(R2)指出“制药开发的目标是设计一个高质量的产品及其制造工艺,以始终如一地提供产品的预期性能”,“质量应该通过设计建立”,而不是通过测试。 此外,它将质量定义为“药物或药品对其预期用途的适用性”,包括身份,强度和纯度等属性(ICH Q6A)。 然而,正如Kozlowski和Swann所述,ICH Q8(R2)及后续指南Q9,Q10和Q11主要关注制造工艺理解,但未将产品知识方面(如产品设计和产品规格)整合到预期用途中。
Juran对质量计划或质量设计的承诺包括建立质量目标,确定客户(利益相关者),确定客户需求(产品应该完成什么),开发响应客户需求的产品功能( 产品设计),以及开发能够生成所需产品的流程(注意这些流程不仅限于制造,如监管链,管理,报销等)。 因此,它清楚地描述了质量设计的核心是满足特定客户要求的产品特征设计,在药品的情况下,其将包括患者以及其他利益相关者(例如,支付者,医疗保健提供者)。
任何新的治疗候选物都需要回答以下问题:是否可以(以适当的成本)进行? 它稳定吗? 是否可以根据预期的给药途径制定? 对患者安全吗? 它能以所需的剂量和所需的时间长度进入所需的组织吗? 它会发挥所需的生物活性并显示出足够的效果吗? 甚至在找到先导候选者之前,可以将这些要求概括为预期的性能概况,并且从该概况中,可以得出有助于确保开发高质量治疗候选者的所需特征。 在这种情况下,可开发性不仅仅涉及制成品的纯度或稳定性方面; 它还提供了一个平台,可以在早期基础上整合产品知识,并从一开始就定义了一个强大的质量目标产品概况(quality target product profile ,QTPP),可以保证成功,安全和有效的药品。
可开发性着眼于给予某一治疗候选药物在三个主要质量领域具有所需特征的不同方面(见表10.1):
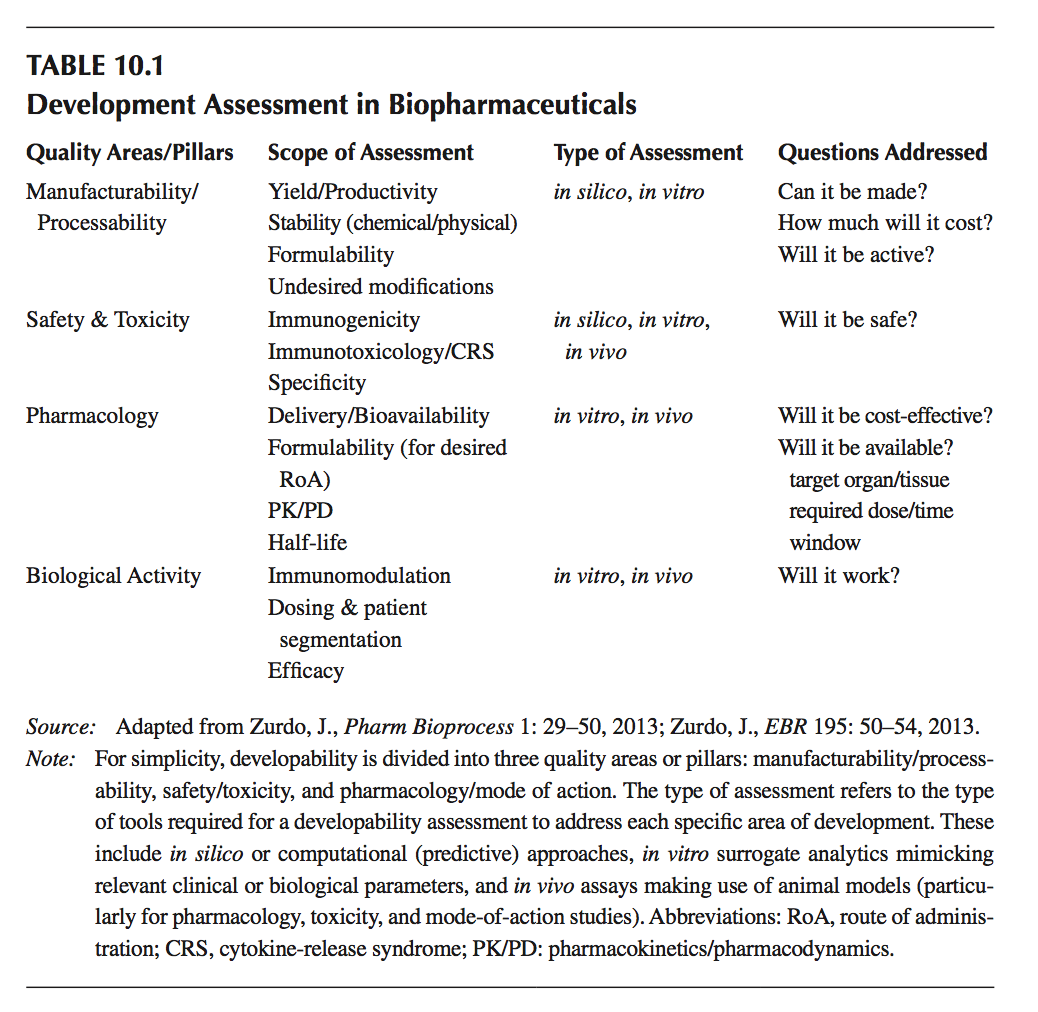
- 可制造性:生产率(产量),产品稳定性,降解和与产品相关的杂质分布,可配方性
- 安全性:免疫原性,免疫毒性,特异性等
- 药理学/生物学活性:生物利用度,半衰期,预期给药途径的配方,免疫调节等
有趣的是,所有这些类别都是相互关联的。 例如,低稳定性会导致聚集,从而导致安全问题(免疫原性), 并且,对于特定的给药途径配制产品的能力确实对给定候选物的生物利用度和药理学(以及通过扩展,功效)具有重要影响。 如需更深入地了解可开发性主题,请访问文献10, 11。
2.2 可开发性:实施整体QBD的三阶段过程
就医药产品而言,对QbD的全面理解将解决产品理解,(制造)过程知识以及两者的相互依赖性。不幸的是,目前医药产品中QbD策略的实施水平有限。 ICH Q8(R2),Q9,Q10和Q11主要处理过程知识及其对产品中关键质量属性(CQA)定义的影响,但没有通过更好地理解产品要求和设计来解决管理质量的可能性产品本身的相关特征。实际上,生物制药生产中QbD策略的传统实施,主要集中在过程验证和过程稳健性上。这种做法绝对有用,但也许忽略了设计产品质量的最重要的一个方面:确定所需的优质产品属性,以及在给定治疗产品的分子结构内设计和优化这些属性的过程。事实上,已经认识到设计药物质量的明显方法是通过设计具有确定的QTPP的最佳分子候选物。
如上所述,可开发性可被视为QbD指导的延伸,在产品知识和过程理解之间架起桥梁,解决产品特性对制造和临床结果的影响,并有助于扩大候选药物的设计空间。 我们展示了如何将可开发性应用于早期降低风险以及如何将其与发现和流程开发活动无缝集成。
从本质上讲,任何可开发性评估计划都包含三个不同的阶段:
1.风险评估。 评估风险的最简单且最具成本效益的方法,是通过使用生物制药候选物序列作为单一输入,来实施能够预测特定可开发性特征的计算方法。 这些方法可以具有非常高的吞吐量并且实现起来相对简单。 在本章的后面部分,我们将介绍一系列可用于风险评估和决策目的的不同工具。
2.实施风险缓解策略:选择替代候选物,重新设计候选物或修改流程。 如果没有适当的纠正措施或缓解策略来解决潜在问题,那么预测潜在问题是没有用的。 根据风险评估过程的具体情况,可以考虑不同的行动方案。 在尚未启动过程开发(即细胞系开发)的情况下,可以探索两种不同的途径:(a)选择具有更好风险特征的备选候选者;(b)重新设计候选者以纠正风险突出的问题
一旦产品已经进入流程开发,或者在无法进行再造的情况下,与流程相关的干预措施可以帮助缓解这些潜在问题。 策略可能包括在细胞系开发过程中筛选大量克隆,以确定那些能够弥补生产力低下,高聚集或稳定性问题的克隆。 其他方法可包括利用替代的下游过程(即,不同的缓冲液,纯化或浓缩步骤)以最小化降解或沉淀的发生,或引入更严格的纯化方案以减少聚集物质的存在。
所有这些策略将在本章后面进一步详细描述,其中包含有关如何以及何时使用这些策略的信息,以及说明如何将风险缓解应用于实际示例的案例研究。
3.确认行动方针。 通过引入适当的验证研究来完成可开发性风险缓解周期。 例如,在生物药物的免疫原性的情况下,可以重新设计候选物以消除序列中特定T细胞表位的出现,然后使用利用来自人类供体的血液样品的相关基于细胞的测定进行测试。
2.3 可开发性工具包:去除早期风险的不同方法
在早期生物制药开发过程中,没有单一的方法或途径可以实现可开发性。 可开发性需要结合使用不同的工具,从使用计算工具到适当的分析方法和测试,以及体内模型,以及尽可能适当的体外替代品。 最后,正如我们将在本章中看到的那样,决策方法对于编制可开发性评估期间收集的信息,并确定其优先级并从中提取有意义的结论至关重要。
我们将描述这些方法中的一些,特别强调计算方法的引入以及特定的体外测定。 但是,我们首先要确定这些不同方法的用途以及它们的局限性。
2.3.1 计算方法
在早期开发中使用计算工具中,由于其相对简单的实施和灵活性而受到越来越多的关注,在高吞吐量,低成本和相对短的分析时间方面提供了相当大的益处。它们也可以在任何给定的时间点应用,因为它们通常不受材料可用性或化验限制的限制。这些方法可以在知道候选物的序列后立即开始构建产品理解。它们提供了一个窗口,这些属性在制造或临床开发过程的许多之后才会出现,并且可以通过选择或设计具有有利特性的候选lead来帮助建立产品质量。目前,有许多计算方法可用于预测生物药物的免疫原性(安全性)和物理和化学稳定性(可制造性),以及其他性质。我们期望在不久的将来,计算方法也将能够帮助设计纯化方案或配方兼容性
从消极方面来看,预测计算工具的功效最终受到其开发中使用的实验信息的类型,数量和质量的限制。这种限制在预期输出由复杂生物学背景(人体)中的多种促成因素(例如免疫原性)决定的情况下尤其重要,所述生理环境在不同种群中也表现出很大程度的变异性(例如,不同的遗传,表型,或患者的疾病背景)。在这种情况下,预测工具通常利用预期属性的替代描述符作为算法的预测输出(例如,T细胞表位含量),其反过来可能由一个或多个促成因素(例如,主要组织相容性复合物(MHC),结合、抗原加工等)影响。因此,重要的是要理解虽然计算工具提供了帮助选择和设计最佳候选药物的非凡机会,但它们还具有内在的局限性,需要通过引入足够的体外和体内评估来平衡,这有助于验证或反驳预测和使用计算机工具的设计。计算方法非常适合检测潜在问题;然而,正如我们稍后将看到的那样,对已识别的风险进行鉴定也很重要(例如,通过考虑结构因素),因为这些方法可能会过度预测并且标记出不适当的大量潜在问题。这是实验性体外或体内验证可用于帮助量化或鉴定实际风险影响的地方。因此,随着开发过程的进展,计算工具对传统的实验方法起到支持作用,有助于指导何时何地可以最有效地应用更昂贵的实验资源。表10.2总结了与计算工具在风险评估中的应用相关的利弊。

2.3.2 代理分析方法(Surrogate/Proxy Analytical Methodologies)
应用有效和强大的早期风险评估中最重要的问题之一,是使用适合目的的适当分析工具和分析方法。通过这种方式,我们不能只移植目前用于晚期产品物理化学表征和稳定性研究的标准分析实践和方法。其原因在于,尽管这些实践提供了高内容和高精度的信息,但它们通常还需要大量的产品,资源和时间。因此,有可能开发出可以将材料需求降低10 ^ 3到10 ^ 4倍的方法,以及将样品通量提高10 ^ 2到10 ^ 3倍。显然,我们不能期望收集与标准分析技术相同水平的信息。然而,在发展的早期阶段,可以快速且廉价地获得的定性信息,例如评估给定分子是否比参考更好或更差,或者在不同候选者的集合中识别最佳分子,可以足以做出决定制定目的并决定哪个lead在发展中向前发展。
在许多情况下,这将意味着替代或代理测定足以定义候选产品的给定性质。早期开发中使用的分析方法正朝着小型化和高通量分析的方向快速发展,并且它们与早期,快速和低成本的分析和计算方法的集成是可开发性概念的核心。例如,评估给定候选物的一般聚集倾向可能不需要高度复杂的评估(即粒子计数,光散射等),但对于高通量毛细管电泳,浊度或间接(代理)免疫测定可能就足够了,例如我们在别处描述的寡聚物检测试验(ODA)。通过实施机器学习方法与高通量自交互色谱方法的组合来确定第二维里系数(B22)作为蛋白质溶解度的替代,已经探索了用于评估高浓度稳定性的高通量策略。同样,许多作者提出通过动态光散射(dynamic light scattering,DLS)和光子相关光谱(PCS)或DLS测量的扩散相互作用参数(kD),在蛋白质样品中使用已知大小的乳胶珠的表观流体动力学半径,作为生物制药制剂粘度的替代品。
Glycoprofiling是产品质量的另一个重要领域,新的分析方法和自动化相结合,以提高样品通量,为有趣的新应用打开了大门。 这些包括提高生物制造细胞系选择的效率或更强大和更快速的工艺设计和开发。 可以想象,在不久的将来,类似的平台将被常规用作早期降低风险的方法,以识别特定候选产品中潜在的质量缺陷
最终,我们想要实施的分析需要适合早期可开发性评估环境中的目的。 因此,给定的分析方法应提供所需的信息量(不少于,不多于),足以进行早期评估,在样本和资源要求低的情况下是经济的,易于以高吞吐量格式执行,并且 快速执行,易于理解。
三、生物药物的可制造性:聚集,化学稳定性和生产力
如表10.1中关于可制造性的三个支柱所述,关键可制造性参数包括生产率和工艺产率,化学稳定性,物理稳定性(特别是蛋白质聚集),配方性(包括高浓度下的溶解度)和不希望的(翻译后)改性。蛋白质聚集和化学降解是制造过程的各个阶段可能出现的两个特别重要的问题,包括发酵,纯化,配制和储存。它们不仅可以影响制造过程的产量和经济性,还可以影响目标产品概况,交付,并最终影响患者安全。从过程的角度来看,通过工艺开发和制造来解决聚合和化学不稳定性可能很复杂,并且会导致成本增加,延长开发时间,并导致有限且昂贵的配方和交付选项。从监管和患者安全的角度来看,生物制药制剂中聚集的发生率本身可构成严重的安全问题,或者是生物制药免疫原性的促成因素。此外,配方和容器内容物与产品的相互作用可能是导致聚集物水平增加的因素,对患者具有潜在的破坏性影响。此外,除了聚集外,化学降解或翻译后修饰(PTM)的发生率也可对生物治疗剂的免疫原性产生负面影响。最后同样重要的是生物制药过敏反应的潜在发生率,这些反应可以通过特定的PTM介导,例如异常(非人)糖基化模式。
3.1 物理稳定性和聚集:计算方法
如前所述,蛋白质聚集体可以存在于不同的发育阶段,并且包含可以通过几种不同机制产生的异质结构组,具体参见Philo和Arakawa综述。 这些包括:
- 天然单体的可逆缔合 (Reversible association of native monomer)
- 构象改变的单体的聚集 (Aggregation of conformationally altered monomer)
- 化学改性产物的聚集(Aggregation of chemically modi ed product)
- 成核控制的聚集(Nucleation-controlled aggregation)
- 表面诱导聚集(Surface-induced aggregation)
本节重点介绍前两种聚集机制(单体介导),而化学修饰产物的聚集将在后面的化学不稳定性和翻译后修饰方面进行讨论。 成核控制和表面诱导(界面诱导)聚集都可以是与过程相关的或与杂质有关的机制,本章将不再讨论。 然而,重要的是要提到虽然各种聚集机制之间存在很大差异,但它们都有一个共同的最终起源:多肽链及其内在的物理化学和结构特性,以及它的化学不稳定性。
多年来,已经开发了许多不同的模型来预测蛋白质的固有聚集倾向,并且其中已经有了许多综述。最早的聚集预测方法之一是由剑桥大学的Dobson及其同事开发的,最初用于成功地将内在聚集倾向预测与溶液中多肽的聚集率联系起来。该模型随后成为AggreSolveTM平台的一部分,与其他工具一起,随着时间的推移逐步发展,将内在蛋白质参数和外在环境因素(如pH和离子强度)结合起来,以解释配方条件的影响。该原始方法使用多肽的氨基酸序列作为主要输入,并且最好用于描述从构象改变,未折叠或部分折叠状态的聚集。然而,该工具的后续进化包含了蛋白质构象稳定性的其他描述符,例如描述了完全折叠状态的聚集倾向。该方法已在生物制药的重新设计中成功实施,以降低其聚集倾向。
当比较高度相似的候选物(即亲本分子的序列变体)的聚集倾向,以及通过蛋白质工程方法检测和破坏聚集热点时,聚集预测算法通常是有用的。然而,在没有具有相似性质的参考蛋白质的情况下评估给定生物治疗剂的聚集风险仍然具有挑战性,其中已知实验聚集性质。当评估具有不同天然构象稳定性的不同类别的蛋白质时尤其如此,这些蛋白质通常与其生物学功能相关。此外,蛋白质在合成,纯化,配制,储存,分配和施用过程中暴露的环境,主要与表达系统的特定成分,产物浓度,pH,剪切力或暴露于气液和空气聚合物界面有关,通常是影响其聚合行为的关键决定因素。例如,给定分子在其最终产品制剂中可以是完全稳定的,或者在给药后在体内在血浆中循环,但在低pH下具有高聚集倾向,这可能在制造过程中呈现特定的挑战(即病毒灭活)。
在任何情况下,许多目前批准用于人类治疗用途的生物药物是抗体或抗体衍生物,并且对其性质有大量知识。与其他蛋白质类别相比,抗体是极其稳定的分子,但它们偶尔也会表现出非常高的聚集水平。事实上,即使在所谓稳定的商业抗体中,易聚集区域的存在也并不罕见。抗体的本质和它们的生物活性所需的高度可变性,通常通过使用新型结构,随机互补决定区(CDR)文库或用于展示和选择潜在结合物的异源系统而扩展,导致了该问题。大自然选择了可被B细胞有效组装,表达和分泌的分子,并针对半衰期和生物学要求进行了优化。然而,大多数现代药物发现平台并非旨在根据此类要求选择候选者,而是针对其目标的结合亲和力。抗体聚集通常与CDR的氨基酸组成相关。在那些报道的支持该观察结果的实例中,有抗IL-13抗体CNTO60752和Dyax hMab-X抗体。当然,这不是完整的。事实上,抗体稳定性也与分子可变结构域中存在的特定框架区相关。特别是一些种系家族通常与更好的稳定性和更高的表达水平相关。例如,已知VH3种系家族是更稳定的重链框架之一,并且与高度可溶的骆驼科动物VHH结构域最相似。此外,有人提出在可变域中存在互补保守相互作用的网络,其显着影响其生物物理特性。
我们小组已经在许多抗体模型中进行了类似的观察,表明可变区中的氨基酸组成(包括框架区和CDR特征)与实验观察到的稳定性和生产力之间存在联系(参见下面的生产力部分)。在此背景下,我们最近开发了一种抗体特异性算法来预测聚集,基于通过在中国仓鼠卵巢 - 谷氨酰胺合成酶(CHO-GS)哺乳动物表达系统中表达数百种抗体获得的实验数据,然后表征它们的差异行为。实验数据集中使用的抗体基于公共抗体序列并选择以覆盖广泛的物理化学和结构空间。机器学习方法用于使用序列和结构描述符作为输入将分子分配到两个不同的类别(低和高聚集风险)。该分类使用实验校准的预定义截止值作为与过程开发和制造期间的聚合事件相关的相对过程风险的指示。该算法在50个不相关抗体的集合中进一步验证,具有良好的可预测性结果。诸如此类的方法是实现高吞吐量和低成本聚合评估的重要步骤,可以将其纳入简单且可操作的可制造性风险中。
3.2 化学不稳定性和翻译后修改
生物制药产品的化学组成的改变,无论是由于细胞过程,酶促,还是化学和降解反应,都可能导致产品微观异质性的复杂水平。据估计,在一小瓶生物制药产品中可以找到多达108种不同的物种。因此,计算方法不应该回答“产品会发生什么样的改变?”而是“可能会有哪些修改发生可能影响其产品性能?“的问题,后者旨在解决应该控制和监控的关键质量属性(CQA)。表10.3包含生物制药开发过程中应考虑的修改清单,但并非所有修改通常都与每种生物制药产品相关。例如,在表中我们突出显示通常在治疗性单克隆抗体中未发现的那些修饰。可以在其他地方找到对不同类型的化学不稳定性和PTM的深入研究。

抗体是最常见的生物制药产品类别之一,并且由于其固有的序列可变性,可以经历广泛的修饰,因此适合于详细讨论的主题。 评估与化学降解和不期望的PTM相关的潜在风险的一种方法涉及使用计算工具来预测它们在特别敏感的位点中的发生,例如被认为对结合亲和力或稳定性重要的区域。 为此,可能影响溶剂可及性,涉及亚基界面或可能与其他溶质相互作用的一级序列和结构约束是要考虑的重要输入元素。 正如我们将在本章后面看到的那样,这种类型的风险评估将用于定义合适的缓解计划。
来自半胱氨酸残基的游离的溶剂暴露的硫醇基团的存在构成了生物制药产品风险最高的化学不稳定性之一。 这些基团的反应性可能潜在地促进蛋白质错误折叠和聚集,以及增加免疫原性反应的风险。 这就是为什么,除非为了功能目的需要,例如在分子中提供缀合位点(即,掺入聚乙二醇或有效负载),优选在进入显影之前除去游离半胱氨酸。 在文献中有几个例子,其中去除这种高风险的硫醇基团对产品质量具有有益的影响。 例如,在抗血管生成素2 mAb MEDI-3617的可变结构域框架中消除游离半胱氨酸导致产物均一性增加,生产力提高26倍,并且熔化温度提高11°C。(替换成什么更好呢?回头看一下这篇文献)
化学品不稳定性对产品性能的潜在影响并不总是很容易确定。 例如,考虑天冬酰胺脱酰胺和天冬氨酸异构化,这是抗体中最常见的两种化学不稳定性。 取决于这些修饰在序列中发生的位置,它们可以对分子的稳定性和功能性具有非常小的影响,或者在严重的情况下,可能导致活性丧失,高产物异质性,并促进聚集和片段化。 这些改性的反应速率受pH,温度,顺序(主要是后续残基的尺寸和氢键能力)和溶剂可及性的影响,异构化以比脱酰胺显着更慢的速率发生。 这些类型的不稳定性可能通过过程控制和配方进行管理,但在某些情况下,由于它们对产品质量的高度影响,它们也可能需要蛋白质工程
甲硫氨酸和色氨酸是最易受氧化影响的两个氨基酸,与它们所处的序列环境无关。 然而,这两个残基的灵敏度部分地由侧链的溶剂暴露决定。 埋藏的残留(Buried residues)不太敏感或需要更长的时间与氧气反应。因此,为了正确评估这两种氨基酸的氧化风险,必须考虑结构信息,包括受影响的残留物在分子中的相对位置。 抗体的Fc和CDR中的氧化事件可能潜在地影响许多CQA(聚集,纯度,同一性),但通常可以通过过程控制来管理
产物异质性和不稳定性也可以是诸如糖基化的细胞过程的结果。正确的糖基化不仅对于赋予给定的生物药物特定的生物学特性是重要的,包括其效力和药理学性质,而且它也可以是产品的充分折叠和组装的决定性因素,并且通常定义其他关键属性,例如然而,在诸如CDR的结合界面中或附近掺入非预期的聚糖结构可能会阻塞产物的结合区域或引入可能对结合亲和力产生负面影响的空间位阻。聚糖结构的支化和组成可以不同,从而引入可能潜在地影响产品特性和其他CQA的进一步的异质性。此外,还值得注意的是,产品中非人聚糖的存在是对生物制药产品的超敏反应和过敏反应的已知风险。尽管糖基改造方法可用于增加稳定性或调节生物治疗的活性,但可能需要补充研究来确定任何给定糖基化位点中过程条件(包括宿主)的影响水平。最后,由于在生物加工过程中与糖(通常存在于培养基中)反应,化学糖化也可以在产品中发生,并且可能潜在地影响一致性,稳定性和免疫原性CQA。可以预测潜在的易受糖基化的位点(即溶剂暴露的Lys)。然而,糖化研究不仅可以更有效地确认易感位置,还可以帮助确定问题的严重程度,以及确定促进或预防其发生的条件。
3.3 产量和表达量
生物制造中经常未被认识到的一个重要方面是生产率或产量与产品稳定性(主要是蛋白质聚集)之间的关系。正如我们之前所描述的,蛋白质聚集和稳定性实际上具有许多不同的面孔。聚集不仅表现为生物加工过程中的浊度,沉淀物或产物损失,而且还可以以细胞内包裹物,低细胞/培养物活力或低生产力水平的形式出现。为什么会这样?生物系统已经发展到有效地处理蛋白质合成,折叠,组装,运输和分泌。为此,他们开发了一系列专门用于防止错误折叠和聚合的工具和系统。遗憾的是,我们尚未能完全适应生物制造中使用的生物平台,以应对行业及其更具挑战性的应用中不断增长的需求。聚集体通常可能通过不同的机制对细胞有毒,并且长期以来已知细胞暴露于蛋白质聚集体或聚集蛋白质的表达可显着影响细胞活力。原核细胞具有形成内含物以分离问题的能力,而这种反应在真核细胞中较少见。此外,分泌细胞,例如生物制药生产中常用的哺乳动物系统,具有非常有效的质量控制系统,其防止错误折叠或错误组装的蛋白质被分泌。当确实发生异常事件时,通常蛋白质在分泌途径中停止并被推向重折叠或降解途径。结果,倾向于错误折叠的产品自然会有较低的细胞分泌机会。我们已经在具有不同程度的稳定性和聚集的多种产品中观察到这种模式。这在哺乳动物系统中尤为明显,并且通常表明高聚集倾向倾向于与低生产率同时发生。当然,我们也观察到例外,这些例外往往与克隆选择偶尔可以抵消待表达的多肽链中包含的内在挑战这一事实有关。抛开异常,我们通常会发现观察到的生产率与观察到的聚集倾向之间存在高度相关性。图10.1显示了在三种不同的单克隆抗体家族中进行的系统分析,这些家族是通过在三种不同的亲本单克隆抗体中引入单突变和双突变而构建的。该图表显示,在每个家族中观察到的生产率和聚集水平之间存在高度相关性。我们还在重新设计抗体以降低其聚集水平时观察到类似的模式(参见本章后面的案例研究)。在所有这些情况下,我们始终看到产品titer大幅增加以及较低的聚集水平。因此,聚集预测可以潜在地用作生产力水平的替代物,特别是在哺乳动物系统中表达的生物药物中。
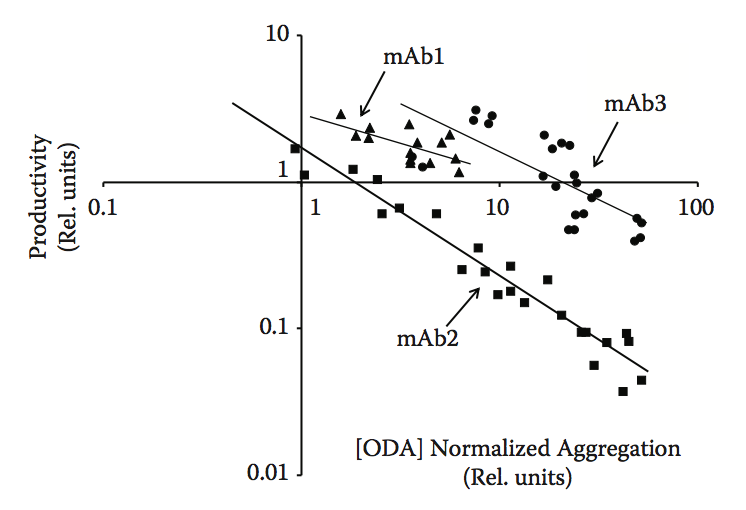
图10.1 分析三种不同抗体家族中抗体生产力和聚集之间的关系:mAb1,mAb2和mAb3。 数据点对应于衍生自三种不同亲本抗体分子的抗体变体(单突变和双突变)的集合。 在相同条件下瞬时表达所有不同的抗体以最小化表达中的任何克隆变异性。 使用Lonza的寡聚体检测试验(ODA)评估相对聚集。 该图表明在每个抗体家族中观察到的聚集和生产力水平之间存在某种程度的相关性。 (Adapted from Zurdo J. et al. Biomed Res Int, Vol. 2015, Article ID 605 427, 2015.)
此外,在治疗性抗体的情况下,我们和其他人已发现抗体分子的特定区域的氨基酸组成与哺乳动物系统中观察到的生产力之间的联系。 这些观察为开发预测平台打开了大门,通过计算工具评估产品表达。
3.4 配方的重要性:公式评估
生物治疗递送正变得越来越重要,不仅因为它对产品药理学的潜在影响,从而影响其功效,还因为它对与患者依从性甚至给予治疗的成本相关的其他重要产品属性的影响。 例如,据估计,在某些极端情况下,与在医院和专业监督下输注生物制药产品相关的成本可能超过产品剂量本身的成本。 这带来了来自付款人的压力,以降低治疗的总成本,同时也通过开发可能有助于自我管理的配方和递送方法来促进患者的依从性。
与传统输注方法相比,皮下输送生物治疗剂具有许多优点。 它简化了药物的给药,侵入性较小,减少了患者的不适,并且可以通过促进产品的逐渐/持续释放来调节产品药理学。 此外,它与自动注射器设备兼容,便于患者自行管理。 然而,皮下剂量限于1.5ml体积,并且通常涉及100至200mg / ml的产品浓度,以便递送足够的剂量。 这种高浓度制剂的使用给产品质量带来了新的挑战,例如溶解度限制,高粘度,聚集体的存在,以及可能阻碍皮下给药途径(RoA)用于递送的相分离。
这是为什么在产品开发过程中对配方兼容性或配方性的良好理解原因之一 给定产品候选物对所需RoA或递送方法的适用性的评估在产品开发过程中特别重要。然而,可配方性(formulability)在其他开发领域也是一个非常重要的参数,特别是在工艺开发和制造方面。例如,大多数开发过程是使用标准方法和溶液(包括色谱纯化,渗滤或产物浓缩)开发的,这些方法和溶液可能与给定的候选产品完全不相容。不幸的是,在产品在给定缓冲液中不稳定或不能耐受特定pH范围的情况下,产品损失是常见的。这也是产品在制造过程中需要集中的情况。例如,在下游过程中,由于植物在给定时间可以储存的体积限制,药物储存可能需要25至200g / L的浓度。在当今的制造平台中可以实现的产品滴度的提高,恶化 (exacerbated)了这个问题。
我们仍缺乏简单的配方平台来评估给定产品候选物在高浓度(即皮下给药)或与基本溶液和工艺条件的相容性时的适用性,以便在制造过程中最大化产品稳定性和回收率。 然而,在这个领域正在开发一些有前景的方法,如下所述,希望其中一些方法能够在不久的将来产生新的可开发性工具。
配方筛选可以通过计算方法在聚集倾向(参见前面),长期稳定性和赋形剂的选择方面进行指导。 然而,目前的配方设计基本上是一个分析学科,其中通常必须考虑多种正交方法的结果,以及随着工艺转移到制造规模的变化条件。 然而,利用高通量方法是筛选宽配方空间的关键,而计算支持可用于支持分析。
已经探索了许多高通量策略来评估高浓度下的蛋白质稳定性。 一种方法依赖于使用第二维里系数(econd virial coefficient)作为通过使用动态光散射(dynamic light scattering,DLS)的蛋白质溶解度的替代物。 此外,许多作者已经提出使用替代参数来通过利用斯托克斯 - 爱因斯坦方程来评估生物制药制剂的粘度。 这些涉及测量蛋白质样品中具有已知尺寸的胶乳微珠的表观流体动力学半径或扩散相互作用参数(kD),由此可以得到溶液粘度。 提出的方法包括DLS和光子相关光谱(PCS)。
即使使用如上所述的相对高通量的方法,待研究的潜在制剂条件的数量也可能是压倒性的。 在这种情况下,使用计算方法可以帮助使筛选过程更易于管理。 一种有趣的方法涉及机器学习计算工具与高通量自交互色谱(SIC)的组合,以估计覆盖非常宽的配方空间(高达数千种不同条件)的蛋白质溶液的第二维里系数,允许设计 生物制药配方,产品可用性非常有限,并且在开发过程的早期阶段。 此外,计算方法也可用于整合来自不同正交分析方法的测量,潜在地允许分析大数据集。 这种方法的例子包括Chernoff面,星图和经验相图。
四、生物医学中的安全:免疫原性和免疫毒理学
与小分子药物相比,生物制药通常被认为对患者更安全。 然而,它们对患者的给药可引起许多不良副作用,通常与药理学问题,作用机制或更常见的免疫原性反应有关。 因此,免疫原性通常被认为是生物治疗药物的主要安全性问题之一,也是早期临床开发过程中磨损(attrition)的主要原因之一。
目前的临床数据表明,大多数治疗性蛋白质具有不同程度的免疫原性。 产生不需要的免疫反应可能对治疗性蛋白质的功效和安全性产生负面影响。 因此,在临床前药物开发期间早期并入免疫原性评估可以显着降低在临床中产生不需要的免疫应答的风险,这可能潜在地改变产品的药理学或使其完全无效。 在极端情况下,生物制药还可能导致严重的超敏反应和过敏或免疫毒性反应,从而使患者的生命处于危险之中。
早期识别免疫原性风险因素还可以采用风险缓解策略(例如,去免疫),从而增加给定产品成功的机会。 在接下来的几页中,我们将介绍一些临床前方法,这些方法可用于评估生物制药候选药物在开发过程中的免疫原性风险及其在各种过程和产品环境中的特定应用。
4.1 需要进行基本的免疫原性评估
免疫原性是物质在人体或动物体内引发免疫应答的能力,可包括体液和细胞介导的反应。 可能需要免疫原性(例如,在疫苗接种期间提高针对病毒或细菌的免疫应答)和不需要的(对治疗性蛋白质的免疫应答,其降低治疗的功效和安全性)。
一旦施用生物治疗性蛋白质,它就会遇到体内许多不同的细胞,包括抗原呈递细胞(antigen-presenting cells ,APC)(图10.2)。 这些抗原呈递细胞(包括树突细胞和B细胞)在内部摄取和加工蛋白质。 衍生自该蛋白质的一些肽可以结合人白细胞抗原(HLA)II类分子(也称为主要组织相容性复合物[MHC] II类)并且展示在抗原呈递细胞的表面上。 然后,这些HLA-肽复合物可以与T细胞表面上的T细胞受体相互作用并激活T细胞应答。 然后,来自生物治疗蛋白的特定T细胞表位激活的T细胞可以与也遇到生物治疗蛋白(通过与B细胞受体结合)的B细胞相互作用,并提供帮助以产生针对生物治疗蛋白的特异性抗体。 从而产生针对蛋白质的不需要的反应。
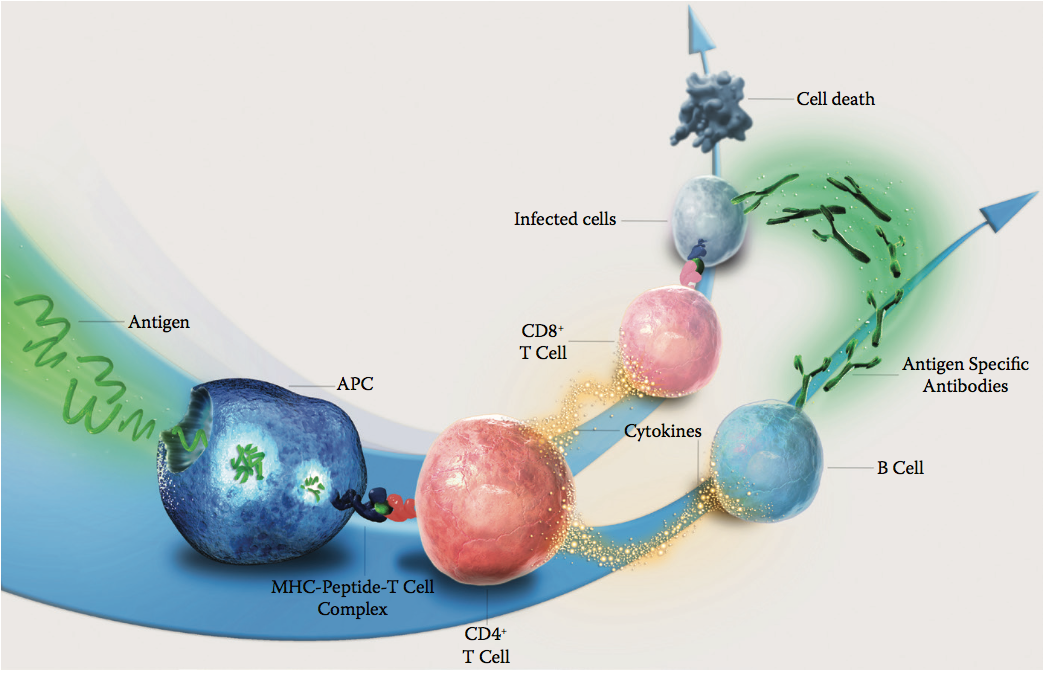
图10.2 示意图显示了引发针对抗原的免疫应答的途径。(Copyright Lonza Biologics plc.)
对治疗性蛋白质的免疫应答是高度复杂的,内在和外在因素都有助于观察到的免疫原性。 内在因素包括蛋白质中B细胞和T细胞表位的存在和非人的翻译后修饰(例如,糖基化)。 外在因素包括配方赋形剂,聚集,降解以及最终产品中存在的任何污染物和杂质。 此外,治疗因素,例如给药途径,剂量方案,治疗时间和个体患者的特征(遗传状态,疾病适应症和伴随药物),也可以影响治疗性蛋白质的免疫原性。
通常,通过监测针对蛋白质产生的抗体的产生,在临床中评估对治疗性蛋白质的免疫应答。 这些体液(即非细胞介导的)反应可以是T细胞依赖性的或独立的。 当B细胞能够识别并结合蛋白质中的表位时产生T细胞非依赖性抗体应答,但是在没有T细胞帮助的情况下,这些通常是低亲和力的瞬时IgM抗体。 当治疗性蛋白质也诱导T细胞应答时,抗体应答可导致高亲和力,长寿命的IgG抗体,其更可能影响临床中治疗性蛋白质的安全性和功效。 由于T细胞反应在长寿命,高亲和力抗体开发中的重要性,因此在治疗性蛋白质的开发过程中重点关注T细胞表位的鉴定和去除,以降低其潜在的免疫原性风险。
有趣的是,监管机构鼓励创新者探索临床前方法的使用,这些方法可以早期指示患者的免疫原性风险,包括计算机和体外方法。 在以下部分中,我们将介绍可用于此目标的一些方法。
4.2 对于基本免疫原性的动物试验的替代方案
目前的监管指南要求在动物模型(例如啮齿动物,非人类灵长类动物)中进行临床前免疫原性评估。 尽管这些模型可用于免疫原性测定的设计和开发,但是人们普遍认为这些非人类模型不能预测临床中可预期的免疫原性水平。 从道德和成本的角度来看,有减少药物研究(例如,英the National Centre for the Replacement, Re nement and Reduction of Animals in Research in the UK)使用动物的动力。 监管机构也鼓励使用全人类临床前技术来补充所需的动物试验,特别是在高风险病例中。
4.3 在SILICO预测免疫原性
如前所述,治疗性蛋白质的免疫原性可以通过许多不同因素来确定。这些可以是内在的(T细胞表位或异常糖基化)或外在的(聚集体,降解产物,污染物,赋形剂等),以及与治疗本身相关(给药途径,给药方案或个体患者的特征) 。 由于免疫反应的复杂性,开发能够整合所有这些变量的预测计算模型将极具挑战性。相反,T细胞表位的存在通常用作免疫原性的替代预测因子。该方法背后的基本原理是B细胞产生抗体需要抗原呈递细胞(APC)呈递非自身T细胞表位。因此,HLA受体可能作为非自身呈递的序列的存在是许多免疫原性反应的先决条件。因此,计算机预测工具集中于鉴定蛋白质中存在的潜在T细胞表位。尽管存在局限性,但计算机筛选在药物设计和先导候选物选择阶段期间提供了高通量且廉价的免疫原性评估方法,甚至在表达和表征分子之前。
在过去的二十年中,已经开发了许多用于预测免疫原性的计算方法。 这些工具中的大多数通过预测来自目标蛋白的肽片段与HLA II类受体的结合特异性来评估蛋白质中的T细胞表位含量。
我们将在此描述用于免疫原性筛选的EpibaseTM内部计算平台,并举例说明其在药物开发早期阶段的应用。 免疫原性风险平台通过预测肽序列与HLA II类受体的结合来鉴定蛋白质序列中的潜在T细胞表位。 该工具使用统计学方法构建,并且基于HLA分子的结构特征,基于序列的肽特征和实验测量的肽/ HLA结合亲和力。 在评估期间,将蛋白质序列分成10-mer重叠肽,预测每种肽的结合亲和力用于HLA II类同种异型,并且肽分类为结合物和非结合物。 在本章的后面,我们还讨论了HLA I类工具,该工具已被开发用于评估表位的免疫原性以支持疫苗的设计。
HLA受体具有高度多态性; 因此,评估给定群体中生物治疗药物的潜在免疫原性风险,需要分析每种构成肽与大量HLA同种异型的结合特异性。 一些同种异型在特定人群中的流行率高于其他人群。 因此,对不同同种异型的群体频率的了解对于选择相关HLA类型和适当评估特定群体中的免疫原性风险是重要的。 为了评估蛋白质的相对免疫原性风险,Epibase平台利用八个主要群体组中的HLA II类同种异型频率和全球群体频率分布。 基于其发生频率选择给定群体的相关同种异型。 全球异型集提供了99%的全球人口覆盖率。
计算机T细胞表位谱分析工具可以在lead选择和优化阶段以三种方式有效应用:(1)根据蛋白质的相对免疫原性风险对蛋白质进行排序,(2)鉴定蛋白质序列中的特定肽 免疫原性风险,和(3)通过帮助去除T细胞表位指导蛋白质再造,这一过程称为去免疫。
为了将蛋白质导联作为其相对免疫原性风险的函数进行排序,需要考虑几个标准。这些包括蛋白质中潜在表位的数量和结合的混杂性等。 HLA-DRB1同种异型通常是免疫原性谱分析的主要关注要素之一,因为DRB1受体的表达水平远高于其他同种异型。此外,可以使用自身肽过滤器来排除来自内源蛋白质中存在的序列基序的假阳性的贡献;可以应用不同的过滤器,这取决于所分析的生物治疗剂的类型。 Lonza的Epibase平台在一个分数中包含许多不同的因素,根据各自的潜在免疫原性风险对不同的生物治疗性候选药物进行排名。图10.3显示了对选择的商业治疗性抗体预测的相对免疫原性风险。正如预期的那样,预测的免疫原性风险对于鼠和嵌合抗体更高,对人和人源化抗体更小。
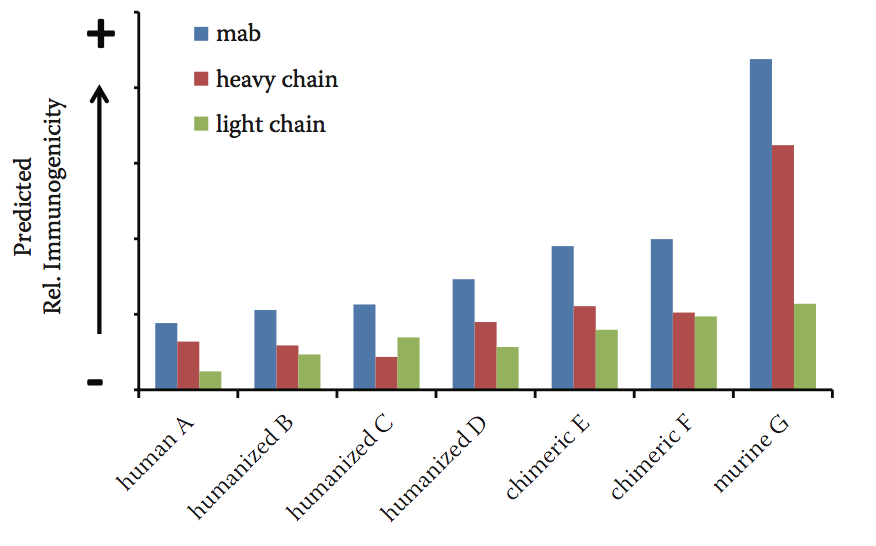
图10.3 预测的一系列市售治疗性抗体的相对免疫原性。
在存在高免疫原性风险的情况下,计算工具可以帮助鉴定潜在的免疫原性肽并通过蛋白质再造来指导T细胞表位的去除。 T细胞表位的计算机制图显着减少了促进分子中高风险肽的选择所需的工作和时间,而不是必须筛选构成蛋白质序列的每个单独的肽。 此外,计算机工具还可用于以非常高通量的方式评估潜在序列修饰对预测的免疫原性的相对影响。 这有助于快速选择可有效降低生物治疗剂的免疫原性风险的最佳修饰。
Epibase平台使用相同的原理对各个表位和整个蛋白进行排序; 肽的得分越高,与该肽相关的免疫原性风险越高。 如果需要去除高风险表位并需要蛋白质工程,则需要进行去免疫热图,如图10.4所示, 提供了一种宝贵的可视化工具,可以快速识别潜在合适的突变,从而降低蛋白质的免疫原性潜力,并结合产品的结构模型。
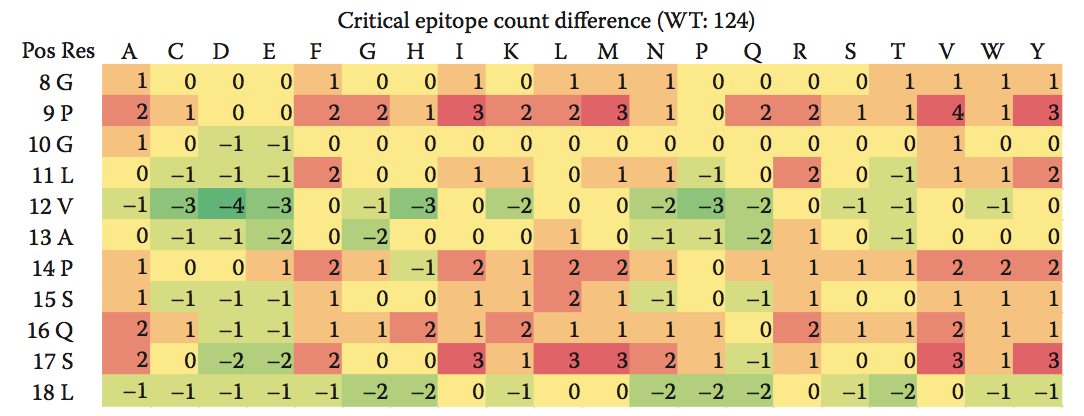
图10.4 去免疫热图显示所有可能的单点突变对蛋白质中关键表位计数的影响。 蛋白质序列显示在最左边的两列中,而每个氨基酸残基的可能取代显示在顶行。 具有负值(绿色)的细胞对应于减少蛋白质表位计数并因此降低潜在免疫原性的突变。 具有正值(红色)的细胞对应于增加蛋白质关键表位计数的突变。
4.4 体外和体外细胞分析
体外和离体基于细胞的分析平台具有能够在完全人体系统中评估和表征对治疗性蛋白质的免疫应答的优点,从而在首次进入人体试验之前提供关于蛋白质安全性的重要信息。 基于人离体细胞的测定平台具有额外的优点,即能够评估不仅仅是一级氨基酸序列的潜在T细胞表位含量。 这些测定还可以包括分析蛋白质样品中存在的任何构象表位(例如B细胞表位),杂质(例如聚集体或颗粒)和污染物(例如宿主细胞蛋白,内毒素)。
用于离体测定的人原代细胞的来源和质量至关重要。获得高活性和高功能的外周血单核细胞(PBMC)将确保始终保持最佳的测定灵敏度和稳健性。根据我们的经验,人类供体的专门血液采集和PBMC的即时,优化分离和冷冻保存对于确保细胞产生一致的结果,以及使用细胞的不同离体测定平台具有所需水平的稳健性是必不可少的。应选择在这些离体测定中使用的供体以匹配预期的目标群体(例如,将密切代表I期临床试验的全球群体)。通常,从健康供体分离的PBMC部分适合于评估T细胞和B细胞对治疗性蛋白质的反应。然而,值得注意的是,PBMC可以来自患有特定疾病适应症或具有可能与正在开发的治疗剂相关的特定遗传或种族背景的患者。例如,PBMC可以来自患有类风湿性关节炎的患者,以评估他们对正在开发用于治疗该病症的治疗性蛋白质的反应,因此考虑到预期患者的免疫状态和遗传背景(即,HLA同种异型makeup)。从具有靶向疾病适应症的患者中取出的PBMC的使用,最终可以更能代表在随后的临床试验中可以观察到的免疫应答的类型。
目前正在使用许多完全人体外离体分析平台来评估免疫原性风险,下面简要讨论一些完整性; 然而,在本书的其他地方更详细地讨论了体外/离体测定平台。
4.4.1 MHC相关肽蛋白质组学
虽然计算机工具能够预测可能存在于蛋白质序列中的潜在T细胞表位,但是它们目前无法准确预测在摄入APC后治疗性蛋白质在内体内被处理的确切方式。 MHC相关肽蛋白质组学(MHC-associated peptide proteomics, MAPPs)测定用于鉴定来自治疗性蛋白质的天然加工的HLA结合肽序列。在MAPP测定中,CD14 +单核细胞从PBMC样品中分离并用GM-CSF和IL-4分化成未成熟的树突细胞(iDC中)。然后收获iDC并加载治疗性蛋白质,然后用细胞因子混合物(通常包括脂多糖(LPS)或肿瘤坏死因子-α(TNFα)/ IL-1β)活化以产生成熟的树突细胞(mDC)。 iDC在蛋白质摄取方面非常有效,而mDC适合于蛋白质加工和通过细胞表面上的MHC II类呈递。 mDC还在细胞表面上表达高水平的T细胞共刺激分子,因此引发与CD4 + T细胞相互作用并激活CD4 + T细胞。这些引发的mDC表达大量的MHC II类分子,其载有来自内源性细胞蛋白和DC负载的治疗性蛋白质的肽。然后可以收获,裂解这些mDC,并通过亲和层析纯化MHC II类(通常为DRB1)分子。随后可以从复合物中洗脱结合的肽并使用质谱法鉴定。然后可以将鉴定的肽与治疗性蛋白质序列进行比较,以鉴定将呈递给CD4 + T细胞的结合肽。 MAPP测定更接近地表示将在体内发生的HLA结合并且与计算机预测相比将降低假阳性率,但是该测定不提供计算机工具提供的通量和成本效益。同样重要的是要注意,计算机模拟和MAPPs工具只能预测治疗蛋白中潜在的T细胞表位, 并且需要随后评估这些鉴定的表位是否能够结合T细胞受体(TCR)并激活CD4 + T细胞。
4.4.2 T细胞活化分析
可以在体外测量T细胞活化的多种生物学结果,包括细胞内细胞因子表达或细胞因子分泌,以及细胞表面活化标记和增殖。 可以通过流式细胞术评估细胞内细胞因子和T细胞增殖,其具有能够标记细胞以表达其他表面标志物(例如,表征T细胞表型,包括Th1,Th2和Th17)的优点。 可以评估在体外T细胞测定期间分泌到上清液中的细胞因子水平,以确定免疫应答的大小和质量。 分泌的细胞因子可以通过多种方法评估,包括酶联免疫吸附测定(ELISA),Luminex或ELISpot。
离体T细胞测定的形式也非常重要,在选择最合适的测定时应考虑许多与产品相关的因素。 这些与产品相关的因素包括蛋白质的性质(例如,肽,抗体,抗体片段,新型蛋白质支架,融合蛋白和重组蛋白),蛋白质的作用方式(例如,毒性或免疫调节蛋白质可能会干扰某些蛋白质)和蛋白质的纯度(例如,一些测定形式对内毒素和聚集体更敏感)。 通常需要优化预期的测定形式以确保最合适的测定形式用于治疗性蛋白质。 优化参数通常包括蛋白质剂量,测定的动力学和测定中的干扰(例如,与阳性对照共培养以鉴定测试蛋白质的任何抑制作用)。
基于PBMC的离体分析可用于评估T细胞对短肽(通常用于鉴定蛋白质序列中特定T细胞表位)或完整蛋白质的反应(以提供对完整蛋白质的免疫原性风险的评估) 。通常,筛选项目包含来自至少50个代表目标群体的供体的PBMC样品(通常是健康供体,但也可以是来自患病患者的样品)。全PBMC测定简单地将PBMC与测试蛋白质以及合适的对照/参照蛋白质共培养,并且在5-7天后通过流式细胞术或ELISpot评估T细胞的活化/增殖。通过耗尽CD8 + T细胞可以增加整个PBMC测定的灵敏度,从而富集CD4 + T细胞群。全PBMC测定通常不适用于调节免疫系统的产品(例如,T细胞靶向治疗剂),其中许多蛋白质抑制T细胞增殖并掩盖任何内在T细胞表位的影响。对于免疫调节蛋白,通常使用基于树突细胞的测定。在这些测定中,单核细胞从PBMC中分离并以与MAPP测定法大致相同的方式分化成树突细胞(参见上文)。 DC加载治疗性蛋白质并被激活,使得蛋白质序列中的任何潜在T细胞表位在细胞表面上表达为HLA-肽复合物。活化的DC还表达高水平的T细胞共刺激分子,因此是能够识别HLA-肽复合物的任何T细胞的非常有效的激活剂。然后将这些活化的DC与自体CD4+ T细胞共培养,并通常通过流式细胞计数监测T细胞活化/增殖。由于使用了有效的抗原呈递细胞,该测定是高度敏感的。
4.4.3 人工淋巴结
正在开发许多平台以在体外更紧密地代表人体免疫系统并试图复制淋巴结的自然环境,包括3D结构和抗原呈递细胞,T细胞和B细胞的比例。 这些模型中的一些还包括添加内皮细胞和流通系统以促进淋巴结样结构的组装。 这些人工淋巴结系统已经产生了关于蛋白质疫苗的免疫原性的数据,并且还可以用于预测治疗性蛋白质的免疫原性。
4.4.4 B-Cell Assays
尽管体外检测原代B细胞反应非常具有挑战性,但检测先前存在的B细胞反应是可能的。对于目前正在开发的许多新型蛋白质治疗剂的预先存在的抗体的普遍性越来越受到关注。许多新型蛋白质支架和小抗体片段正在被修饰以延长分子的半衰期。一种这样的半衰期延伸技术是PEG化,并且最近的报道显示高达20%的健康一般人群具有可检测的预先存在的针对PEG组的抗体。一些新型抗体支架还报道了临床中预先存在的抗体的问题,导致显着的延迟和与鉴定B细胞表位和重新设计分子相关的增加的成本。体外B细胞测定可潜在地用作临床前开发期间的筛选工具,以提供对治疗性蛋白质的预先存在的抗体的流行性的评估。
为了评估预先存在的B细胞对治疗性蛋白质的反应的流行,来自健康个体(或来自患有疾病适应症的患者的样品)的PBMC样品,经历多克隆刺激以将记忆B细胞分化为浆细胞。 然后可以通过B细胞ELISpot评估这些浆细胞,以确定它们是否产生与被评估的治疗性蛋白质交叉反应的抗体。 筛选预先存在的B细胞应答可以在先导选择期间,应用于选择具有最低临床免疫原性风险的候选物。
已知对一些生物药物的不良反应,可与识别产物中存在的非人表位的预先存在的抗体(IgA,IgM,IgG或IgE)相关联,例如非人乙二醇表位。 有趣的是,已经提出至少一些对生物治疗剂过敏的病例可能与治疗开始前存在能够与产物反应的IgE抗体相关。 所有这些都表明,这种类型的分析可能有助于在进入临床前避免过敏反应(hypersensitivity)或过敏(anaphylactic)反应。
4.5 聚合与免疫原性
正如本章前面所讨论的,聚合是在治疗性蛋白质的可开发性评估期间要考虑的重要产品质量属性,以确保蛋白质是可制造的。在聚集和免疫原性之间也存在充分记录的联系,蛋白质的聚集形式诱导比蛋白质的单体形式更高的免疫原性。因此,本章前面讨论的减少聚集的许多概念也将降低免疫原性的风险。然而,大多数治疗性蛋白质含有至少低水平的聚集体,并且目前还不知道聚集的类型和量可以增加免疫原性的风险。临床上有一些例子表明,聚集水平的增加与免疫原性有关。 Eprex®是一种人类促红细胞生成素(EPO),经历了一项配方变化,随后与内源性EPO形成的抗体形成增加有关。这种增加的免疫原性与纯红细胞再生障碍(PRCA)有关。随后的研究表明,这种配方的改变可以促进EPO分子结合并显示多聚体表位的胶束的形成。另一个例子是IFNβ1a,其中有两种产品可用于临床治疗多发性硬化症(MS):Avonex®和Rebif®。 Avonex似乎诱导低水平的免疫原性(~2%),而Rebif在MS患者中具有高免疫原性(~25%)。在这种特殊情况下,观察到的免疫原性率可与每种产品中的聚集水平相关联,Rebif表现出比Avonex更高的聚集水平。最近的数据也表明,单克隆抗体的聚集可导致体外树突状细胞表面HLA II类分子呈现的潜在T细胞表位发生显着变化。
驱动免疫原性和聚集的因子通常位于治疗性蛋白质的相同区域(例如,在单克隆抗体的CDR中和周围)。 在选择铅lead选物或工程分子进行lead优化时,考虑对多种因素的影响非常重要。 有趣的是,最近的一些研究表明,至少在一些生物治疗药物中,可能存在T细胞表位与聚集倾向区域的一些共定位,后者主要受相对大量疏水残基的存在的影响。
4.6 临床免疫毒理学
目前开发靶向免疫系统组分的治疗性蛋白质(例如免疫细胞或细胞因子)的趋势,也增加了过度刺激免疫系统并导致对治疗性蛋白质的免疫毒性反应的风险。这在抗CD28单克隆抗体TGN1412的首次人体试验中清楚可见,其中诱导了严重的炎症反应,其包括细胞因子释放综合征(CRS)(“细胞因子风暴”)和多器官衰竭。随后的研究表明,导致CRS反应的是CD28激动剂活性,而不是TGN1412的制造,配制,稀释或给药中的任何样品污染或错误。在这种特殊情况下,体外和体内的临床前研究未能预测CRS的诱导,主要是由于体外使用人PBMC的次优条件和体内人和灵长类动物之间免疫系统的差异。最近,目前已经开发了许多似乎能够检测CRS反应的新的体外/离体测定,因此已经提出作为在治疗性蛋白质的临床前开发期间评估这种类型的风险的新工具。
4.7 免疫原性和疫苗
在亚单位疫苗,免疫疗法和诊断学的开发中,免疫应答的启动是需要和必需的产品属性。这些病例的目的是通过激活细胞毒性T淋巴细胞(CTL)和T辅助细胞(Th)来鉴定可以诱导针对病毒或癌细胞的保护性免疫的病毒蛋白或人肿瘤抗原的区域。 CTL识别由HLA I类分子呈递的肽,并且Th细胞识别由HLA II类分子呈递的肽。为此目的,计算工具可用于通过预测肽与HLA I类和II类分子的结合来鉴定病原体或靶蛋白质序列中的潜在CTL表位和Th表位。合适的免疫原性肽的发现可能涉及筛选整个病原体蛋白质组,这可能使得实验研究在时间和成本方面不可行。在这种情况下,唯一可行的替代方案是应用计算机工具,其可以在短时间内筛选大量序列,以选择预测为潜在表位的肽。然后可以在基于细胞的体外研究中证实这些表位。 Epibase平台可预测对HLA I类同种异型的结合亲和力,为全球91%的人群提供覆盖率。与HLA II类工具类似,Epibase I类平台还将许多参与参数(表位的表位结合亲和力,受影响的同种异型等)组合成单个评分,然后可以根据其预测的相对性对肽免疫原性进行排序,并选择具有最高免疫原性潜力的那些表位。
五、可开发性和决策工具
可开发性评估可被视为质量设计(qualty by design,QbD)方法的真正综合方法。 正如我们在前面部分中看到的那样,可开发性不是列出可能对给定治疗候选物产生负面影响的每种类型的问题。 它既不是衡量每一种能够定义给定产品的特性的特性,也不是用于表征每种可能的杂质,它的稳定性以及它在每种可想象的条件下随时间的演变。 可开发性是指通过根据患者所需的特性和预期用途,确定特定产品失效的最显着潜在原因来评估和管理风险,然后对这些关键风险参数实施适当的控制和纠正措施。
4.1 质量风险管理:在不确定的情况下做出决策
许多科学家在提出可开发性方法时提出的两个关键问题是“我们应该测量什么?”和“我们如何将多个信息组合成单个输出或决策?”在这种情况下,值得一提的是采用大量可用信息(通过测试也称为死亡),通常会阻碍强大的风险评估决策过程的实施。这可能是数据丰富/知识贫乏综合症的明显案例。我们之前介绍了早期可开发性风险评估的新范例,该模式利用计算方法和代理或代理分析来提供相对较大规模的特定风险参数信息。对于这种类型的评估,可用信息非常精确并不重要。但是,必须能够轻松获取所需数据(在时间,材料和成本方面),并且它们提供有关特定关注领域的充分信息。换句话说,数据提供了足够的深度和上下文以使得能够做出决定。
质量风险管理要求在开发过程中采用结构化的风险管理方法,以促进决策。 这听起来很简单,但实际上实施起来相当复杂,特别是当其应用处于多学科和复杂环境(如药物开发)的背景下时。 我们并不是假装我们只会在几页内解决这样一个难题; 但是,在下面的部分中,我们举例说明了可用于早期可开发性评估策略的思维和决策工具的类型。 我们希望这将促进未来的进一步讨论,并希望制定旨在为更简单和更有效的生物制药发展提供指导途径的举措。
5.2 理解因果关系
尽管量子力学和相对论的发展在二十世纪早期篡夺了牛顿力学的假定精确性,但对于复杂的元素,我们在强烈的因果关系假设下运作; 也就是说,对于每种效果,都存在(或是)明确的原因。 由于在解决问题,设计实验和流程优化方面取得了数千项成功,我们明确地使用了基于因果关系的结构化问题解决机制。 任何结果(预期的或意外的)都是由于最终导致结果的初始条件而发生的。
5.2.1 理解多因素语境中的因果关系
在涉及许多促成因素的复杂过程中寻找因果关系,引入了因子相互作用的更大概率,因此使用轶事,观点或启发式方法来猜测结果的能力要低得多。 尽管这些复杂的过程可能会逃避对其结果的猜测,但它们无法回避经验科学。 这是在一个因素(one factor at a time,OFAT)上切换到实验设计(design of experiments,DoE)的基础。 DoE允许阐明对单个或多个结果的几个同时因素影响。 交互因子将被捕获,并且通过简约原则将消除模型中的微不足道的因素
做出偏向个人启发式(并远离证据和数据)的决策的倾向在行业和我们的整个生活中无处不在。 决策应该有利于将风险降低到实际可达到水平的情况,如果可以做出多种选择,那些路径中的决策质量最高,风险最低(假设每条路径的最终结果相同)。
我们在这里提出的决策制定的最佳方式是一个结构化的思维过程,通过该过程,解决方案的推导本身是以一种正式和可重复的方式完成的,而不是一个试错过程,最终成本更高,风险更大。 从基础智力和经验主题专业知识的结合,我们做出影响多因素系统的决策 - 这些启发式决策实际上对它们没有可重复性,因为每个决策者都是按照自己的预设偏差这样做的。 由于使用适当的风险管理工具具有显着的潜在能量,个人通常会默认其内在的系统偏差。 这带来的问题严重程度包括巨大的财务,时间和劳动力相关成本。 非常重要的是,我们在以下部分提供以下风险评估和问题解决工具列表,以及它们的使用原则。
5.2.2 因果模型:鱼骨图 (Cause-and-Effect Models: Fishbone Diagram)
因其形态类似于鱼骨架而得名,所以鱼骨图在大多数强大的问题解决和质量计划中无处不在。它代表了质量的七个基本工具之一,并且是质量改进大师Kaoru Ishikawa(解决问题的鱼骨图的发明者)的基础,他说,巧妙地使用七种质量控制工具将解决95%的工作场所问题。在图10.5中,我们显示了一个典型的鱼骨图,对应于散装产品中高聚集水平的问题。在该图中,鱼的每个肋骨代表了潜在原因的主要类别,导致研究中的最终效果。在收集到足够数据的情况下,概率也可以归因于鱼的肋骨,以表示一种因果因子与另一种因果关系的相对可能性。通过这种方式,可以在视觉上将问题(鱼的头部)解构为其因果因素,以突出需要进一步调查和改善的部分。
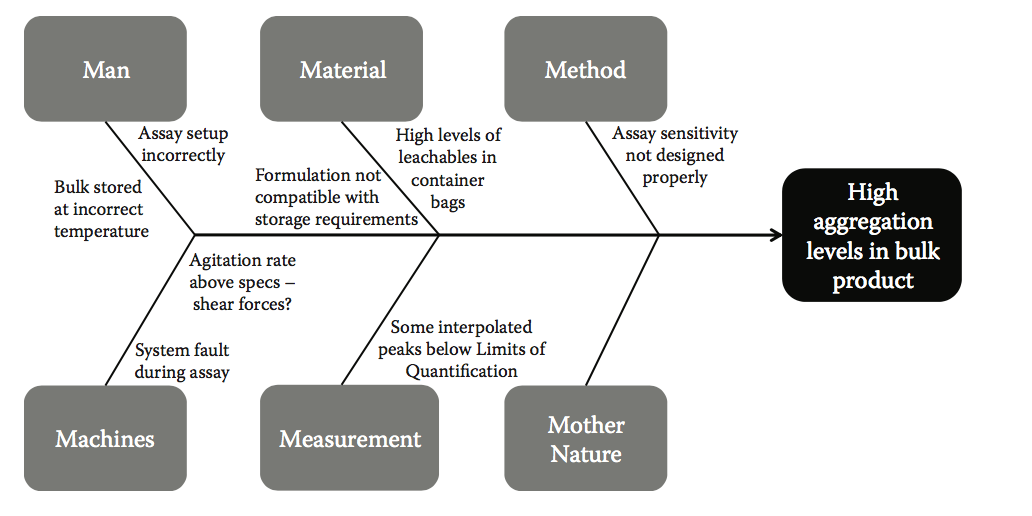
图10.5 鱼骨图详细说明了问题陈述(散装产品中发现的高聚集水平),以及可能导致观察到的失败的主要因果类别和子元素。
5.2.3 五个为什么
如果可以在鱼骨图上捕获不同的项目,那么这五个是一个递归工具,可以在一个特定的故障模式中深入研究。 例如,五个问题可能如下:
- 问题:样品的pH值反复不在范围内
- 为什么? 从一位分析师到另一位分析师的样本未做类似准备
- 为什么? 分析人员使用不同的移液器进行相同的制备程序
- 为什么? 标准操作程序(SOP)没有指定一个移液器与另一个移液器,允许测量变化
- 为什么? 标准操作程序不是与地区主题专家一起编写的
在这种特殊情况下,四个原因足以导致因果因素的特异性,包括或接近根本原因。 提到五个为什么是乔治米勒的“神奇的七号,加号或减二号”的遗产,并不一定要按字面意思; 它只是指在第一个原因之后不停止。 有时确定根本原因可能需要更多,因为它完全取决于流程。 此示例还指出了其他潜在的系统性问题(为什么QA或文档部门允许在没有主题专家的情况下编写SOP?为什么实验室或校准组允许将不同的校准测量设备[移液器]带入测试空间?)
5.2.4 使用图表进行因果关系分析
图形工具(图表)可用于帮助决策过程,通过突出质量偏差或识别瓶颈,可变性的潜在原因,影响所需输出的主要因素等。 下面我们提一些这些工具作为介绍; 有关不同类型图表及其用途的详细信息,请参见其他地方。
- 帕累托图:帕累托图表显示某一部分原因在不同程度上导致结果的程度。许多帕累托图表只显示影响优势效应的几个因素,以突出那些更重要的因素。有人称之为规则。
- 流程图:流程图描述了流程应如何理想运行。流程图可用于突出显示与理想工艺流程的偏差并快速修复它们。
- 直方图:直方图用于评估不同特征(箱)的观察频率分布。分布的形状,中心和范围可以帮助识别异常并支持进一步调查潜在原因或失败模式。
- 运行图表:运行图表是线性图,显示单个变量随时间的变化,用于快速识别可能有助于采取纠正措施的偏差(最好是实时)。
- 控制图:控制图是过程控制中常用的运行图的变体。通常,控制图包括中心线(平均值)和上限和下限。控制限制基于历史数据计算,并定义给定参数或可观察的可接受水平。它们通常用于定义流程何时在规范内或规范外运行,并确定需要进行额外调查的偏差。
5.3 允许决策制定的多参数排名方法
正如我们在本章前面提到的,成功的生物药物需要满足多个标准。 当然,它需要显示出足够的生物活性和效力,但它还必须符合其他要求,例如其以可接受的产率和成本制造的适合性,或者为其预期的给药途径配制。 此外,它在储存和给药期间应该足够稳定并且对患者是安全的。
在这种情况下,药物开发者必须面对的最重要的任务可能是候选者的选择和优化,以纳入最终的特性,这些特性最终导致他们作为治疗性治疗的成功。 这需要实施评估和决策工具,以便根据多个标准对治疗候选物进行排名和优化。
在小分子药物发现领域,已经开发了许多计算工具和方法来基于多种性质对化合物进行排序和优化。然而,在生物药物开发的情况下,使用类似的工具 推动决策过程仍然处于起步阶段,或者在很多情况下,这在很大程度上是闻所未闻的。 也许这些差异的一些原因在于缺乏针对许多生物药物特性的强大的计算预测工具,这些工具可能与其作为治疗剂的成功相关,以及与小的分子相比,生物制药开发中计算工具的相对较少的吸收。 例如,定量结构 - 活性关系(QSAR)方法是小分子药物发现和开发的标准实践,而它们在生物治疗药物中的应用充其量是有限的。
有许多元素可以对多参数排名和优化方法产生重要影响:
- 所需结果中不同特性的相对重要性/重量
- 要考虑的不同属性的所需数据范围(或阈值)
- 可用数据的不确定性或可靠性:模型的可信区间,分析准确度或先验知识
- 多个值来描述特定属性,例如,计算机和体外数据
下面我们将介绍多参数优化中使用的一些常用策略,以及这些不同元素如何构成影响所做决策质量的特定挑战。
5.3.1 连续过滤
用于决策和候选选择的最常用方法之一是连续过滤(consecutive ltering)方法。 这种方法背后的想法是一次只考虑和优化一个属性。 确定属性或期望值范围的合适阈值,然后将满足该标准的铅分子推进到后续阶段。 这种方法的一个例子是根据其活动对候选药物进行早期筛选和选择。 连续过滤策略的常见问题是一种特定性质(例如,结合亲和力)的优化可能使其他期望的性质(例如,产量,免疫原性或聚集)变差。 如前所述,需要更先进的策略,以允许在药物发现过程的早期进行多参数分类和优化。 使用in silico工具实现多个属性的分析将有助于解决此问题。
导致过滤方法不利的另一个因素是,在采用硬截止时,通常不考虑数据中存在的不确定性。所有类型的数据都存在不确定性:实验室测量受到实验误差的影响,而模型精度将影响 计算机预测。
5.3.2 加权和
多参数优化的另一种常见方法是使用对应于不同属性的排名值的加权和来获得主要候选者的平衡或加权排名。 例如,不同的可开发性属性可以组合在给定分子n(Dn)的客观可开发性排序函数中,其可以采用以下形式:
$$D_{n} = - w^{t} * T_{n} + w^{a} * A_{n} + w^{i}*I_{n}$$
其中wt,wa和wi是反映不同可开发性质的相对重要性的权重因子,Tn,An和In分别是滴度,聚集和免疫原性的相应性质的比例值(测量或预测)。 (所考虑的数据集中的属性值的标准偏差通常被视为缩放因子)。 这三种属性的最佳分子排名最小; 滴定性质的负重量反映了这样一个事实,即我们希望最大化分子的滴度,同时降低聚集和免疫原性。
该方法需要先验地分配权重 –对于不同属性的相对重要性。 排名结果也可以对分配给权重因子的值非常敏感。 权重的轻微变化可能导致不同的排名顺序和不同的最优解决方案。 此外,为不同属性分配相对重要性或权重的任务需要人为干预,因此,它可能是非常主观的。 最后,这种方法没有考虑数据的不确定性。
5.3.3 帕累托优化(Pareto Optimization)
帕累托优化方法可以帮助探索多参数情况,当优化一个给定的属性可能使另一个属性更差。 该方法可以识别Pareto最优线索,并在多个属性之间进行最佳权衡。 图10.6从两个维度展示了这个概念。 理想情况下,目标是最大化两种性质的所需值(即理想分子位于右上角)。 但实际情况是,通过增加属性1,属性2会减少。与其他条件相比,Pareto前端(实心圆圈)上突出显示的点代表具有优越组合属性的条件(empty circles)。 这些帕累托最优点在所分析的两个属性中的至少一个中更好,而在另一个属性中它们并不差。 该方法通常提供许多帕累托最优条件,从中可以由开发科学家选择最佳条件。
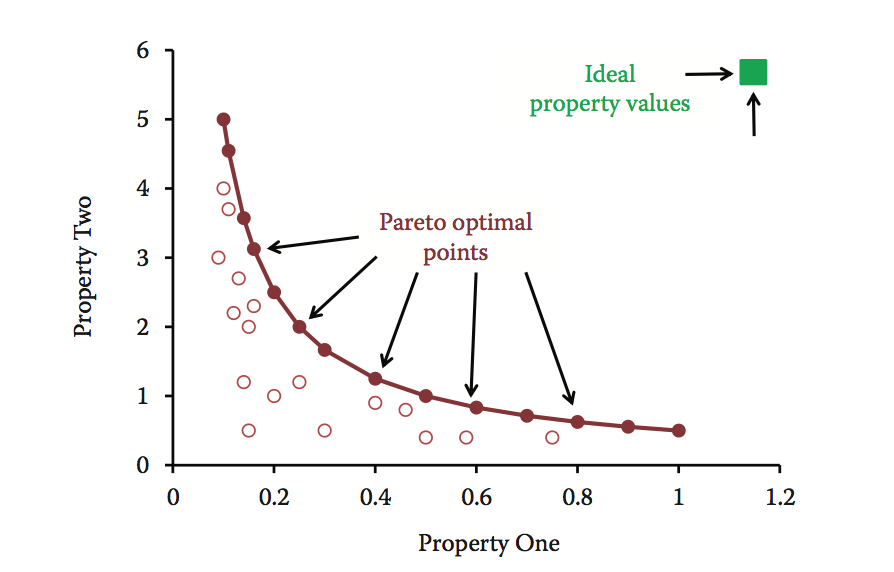
图10.6 两个不同属性的Pareto优化示例。 Pareto-optimal点(Pareto前面)显示为实心圆,非最佳点显示为空心圆。 绿色方块表示理想的属性值。
帕累托最优化方法最适合只需要优化少量属性的方法。 优化期间要考虑的可能点数随着属性数量的增加而显着增加。 当然,在仅需要评估有限数量的分子的情况下,这可能不是问题。 此外,帕累托最优化不需要对属性进行任何先验性的重要性权重分配。 帕累托优化已应用于药物开发,以指导具有最佳吸收,分布,代谢,排泄和毒性(ADMET)特性的小分子的设计和选择。
5.3.4 在决策中纳入不确定性:FMEA和关键性
上述多参数排序和优化的方法都没有考虑所有类型数据中存在的不确定性。 为了解决这个问题,已经开发了包含实验和计算数据,并考虑到数据中的不确定性和各个属性的相对重要性的概率评分方法。 这种方法已用于小分子药物发现,以优化ADMET药物谱
失效模式和影响分析(Failure modes and effect analysis ,FMEA)构成了QbD和质量风险管理(quality risk management,QRM)中使用的大量定量和半定量风险评估原则的基础。 FMEA通过乘法乘积测量过程子元素的假定风险,方法是根据风险严重程度(S),每个风险要素(O)的发生概率和(缺乏)每个要素的可检测性对每个项目进行评分(D)。 这些因素的乘积通常称为风险优先级编号,或简称为RPN(risk priority number)。 然后,针对不同风险因素的RPN通常从最大总体风险到最小总体风险进行排序,以便确定其原则顺序并相应地定义风险缓解策略。 因此,努力和资源的应用可以成为解决那些带来最高风险的因素的优先事项,而减少的努力和注意力则集中在对流程或系统具有较低风险的那些要素上。
$$ RPN = S * O * D $$
编号范围是任意的,具体取决于具体的应用程序和可用信息的级别。 例如,较小的比例(例如,1-5)可能无法提供足够的临时分辨率以能够区分风险或在何处应用分辨率。 此外,如果没有足够的数据来证明给定的线性连续间隔标度,例如1-10,则可以使用使用非连续值的标度,例如1-5-10,低 - 中 - 高或低 - 极少数情况下很少有数据可用。 在任何情况下,该量表的主要目的是促进识别那些实际构成相关风险的特定项目,这些项目值得采用纠正和预防措施(CAPA)。
在确定生物制药开发中的关键风险时,已成功实施FMEA方法的变化。 通常,使用关键性排序代替FMEA,特别是在开发的早期阶段,在只能在过程结束时检测到故障的过程中,或者在这种检测不允许实施适当的控制回路的情况下, 这样可以避免偏离所需的质量规格。 在这样的事件中,风险等级通常在没有可检测性参数的情况下完成。
在生物制药开发中,QbD的应用提供了一个示例,说明决策策略如何整合各种类型的数据以及数据背后的不确定性,特别是在风险评估的实施中。 A-Mab案例举例说明了在人源化单克隆抗体A-Mab的生物制药工艺开发中使用两种不同的风险评估工具。 这些工具可以评估几种质量属性(QAs)的关键性,包括聚集,糖基化,浸出蛋白A,氧化和脱酰胺等,并评估此类QAs对功效,PK / PD,免疫原性和 安全。 为了完成关键性评估,这些工具利用各种类型的可用信息,包括过程实验数据,类似产品的先验知识或临床数据。
工具1:风险排名工具。 其中一个工具根据其影响和不确定性来定义属性的关键性,代表信息的相关性(例如,文献数据或临床研究),用于评估影响:
Criticality (Risk Score) = Impact × Uncertainty (10.3)
影响反映了对安全性,药理学和疗效的已知或潜在影响,而不确定性反映了用于分配影响等级的信息的相关性。 将风险分解为促成因素,针对每个个体因素(免疫原性,功效等)单独评估属性的分数,然后将最大分数作为属性的总体临界分数。
工具2:QA关键性排名工具。 第二个工具根据严重性和可能性得分定义了关键性:
Criticality (Risk Priority Number [RPN]) = Severity × Likelihood (10.4)
严重程度与影响相似,但主要基于先前知识和产品特定信息,并且它考虑与患者安全性,药理学和疗效相关的风险。 可能性估计由质量属性引起的不良事件影响安全性或有效性的概率,并且基于临床,非临床和相关文献信息。
风险评估产生的关键性评分用于定义A-Mab过程开发的关键质量属性。
虽然A-Mab案例研究不是跨多个属性对候选物进行排名的方法,但是使用不确定性的案例研究的一些想法,属性的潜在影响以及不同属性的相对重要性可以扩展到 用于潜在客户选择的多属性排名工具,我们稍后会看到。
5.4 从多参数风险分析角度探讨可行性
在本章的前面部分,我们通过使用不同的工具(计算和分析)说明了如何在不同的开发阶段引入可开发性评估。 下面,我们简要介绍如何使用此类风险评估以及如何实施缓解策略。 我们要强调的是,没有单一的可开发性风险管理工具; 相反,与上述方法不同的方法可以组合在一个风险管理过程中,该过程应该适应特定的产品要求或发展阶段。
例如,控制图可能突出显示制造过程的给定部分中特定化学降解事件(即脱酰胺)的相对较高的发生率。 如果此事件易受产品功效或安全性影响,则可能在FMEA或关键性分析期间触发红旗。 根据上述基本原理,必须通过综合评估其潜在影响或严重程度(S)(即从该事件得出的制造,生物或临床后果)来平衡这种降解事件的发生概率(O)。 如上所述,两者的组合可以提供对这种特定事件的关键性的度量,并且因此建议是否建议为该事件引入校正的行动过程(缓解策略)。 这种方法非常适合QbD策略中使用的传统QRM工具。
Criticality (RPN) = Probability of Occurrence (O) × Severity (S) (10.5)
图10.7中的示例显示了如何根据发生的概率及其相对影响评估不同的参数或事件,并将其用作整个分子的风险指数的一部分,或单独评估以定义缓解计划(需要采取缓解措施)。
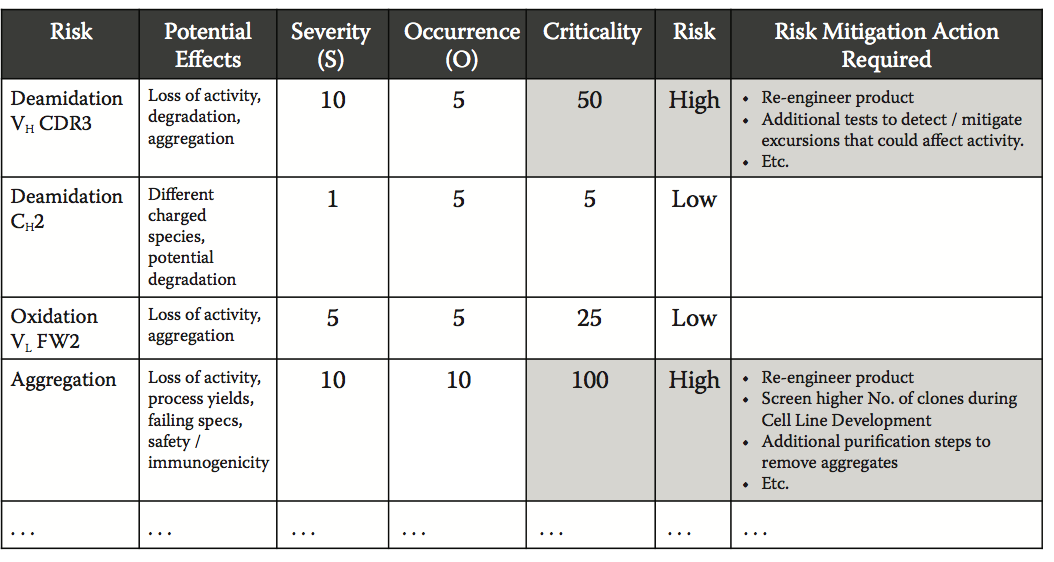
图10.7 单克隆抗体临界评估的实例。 根据不同事件的相对严重程度(S)和发生概率(O)对不同事件进行分类和评估。 为简化评估,严重程度和发生次数均分配以下风险参数:1 =低风险,5 =中间风险,10 =高风险。 在这种特定情况下,出现对应于通过计算机方法确定的相对出现倾向。
或者,可以使用加权函数或帕累托最优化来组合不同的风险参数,以产生对给定风险的更相关的评估。 一种可能的方法可能涉及通过将每个单一风险评分因子定义为由各种其他元素贡献的函数来修改先前提出的加权和模型。 例如,方程10.1中定义的免疫原性评分In可以构建为多种促成因子的函数,例如T细胞表位含量,聚集倾向和化学降解倾向。
5.5 决策可以是产品特定的
我们在本节中要强调的一个方面是,对于可开发性风险评估而言,没有一个适合所有方面的事情。 正如我们之前所讨论的,治疗药物的预期用途,其目标人群,甚至可用的报销制度都可以对产品的设计和制造方式提出具体的限制和要求。 通过这种方式,不同的质量属性将与特定的风险概况和风险管理策略相关联。 因此,重要的是要在一开始就理解这些要求究竟是什么才能确定合适的风险管理方法。( 哈哈,这不就是项目思维么?需要设定好预期目标!! )
在以下部分中,我们将说明在为特定产品定义可开发性计划时如何整合不同的考虑因素,这些产品具有由疾病状况,目标患者人群,商业要求,期望的临床结果,使用的平台和公司定义的特定策略等等。
六、生物制药的可开发性应用:一些例子
6.1 何时以及如何使用可开发性风险评估
如上所述,如果从发现的早期阶段开始应用可开发性方法确实具有强大的有益潜力,确保将正确的质量属性设计到所选择的开发候选中。 在单克隆抗体的情况下,选择最佳候选物可以,例如,开发合适的显示库(display library),其富含有利的生物物理特性(即,化学不稳定的位点和易于触发聚集的区域被移除,或具有T细胞表位 内容减少),可以很容易地转换成完整的抗体格式。 此外,有许多新型抗体平台利用人源化啮齿动物或人B细胞文库或甚至直接从患者分离的抗体。
6.1.1 Lead发现
上述发现源的共同点是它们产生了大量候选者和大量数据。在此阶段使用的最有效的可开发性策略是计算方法,其自动分类或标记高风险候选者并帮助指导在早期阶段选择更有利的分子。例如,前面描述的抗体聚集预测工具与潜在化学不稳定性和Epibase免疫原性(基于T细胞表位)评分的检测结合使用,是早期将物理和化学稳定性与免疫原性风险相结合的一种可能方式。以简单的方式,无需进行任何额外的实验。事实上,随着候选产品进入开发阶段,体外和体内数据的数量不断增加,大多数计算机数据和预测在早期的lead发现阶段已经可以获得(参见图10.8的原理图)。这两个数据来源(计算机,体外)是互补的,但随着项目进一步进入临床前开发,计算信息应该补充或转向更多经过实验验证的数据, 由于所需的吞吐量较低(即,达到这一点的候选物数较少)。
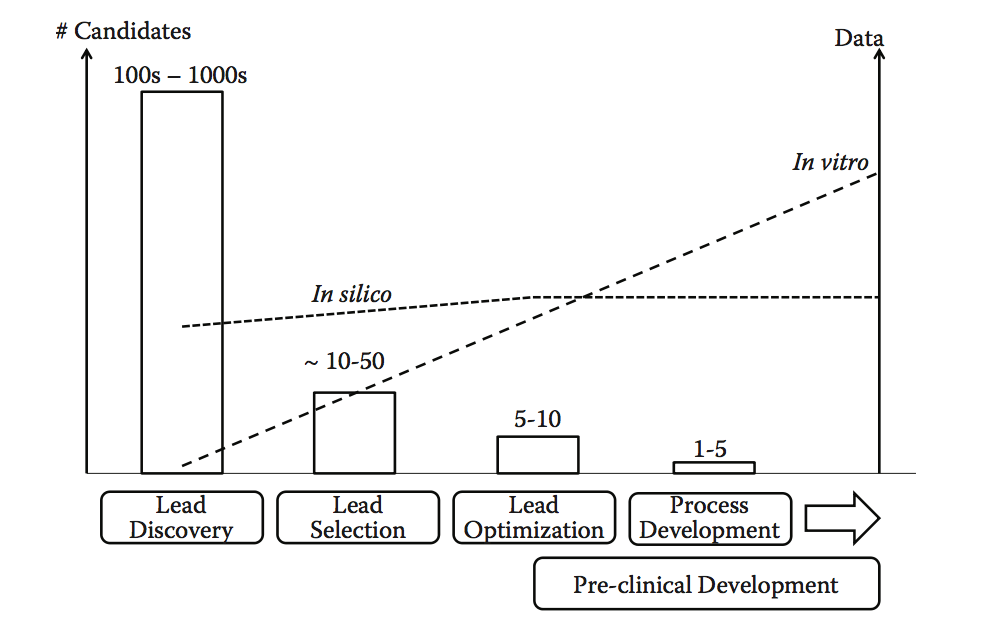
图10.8 在发现和开发的不同阶段实施可开发性方法,以及它们与主要候选者和可用数据的数量之间的关系。 随着潜在候选者的数量收敛到较小的数量(可能是单个lead),可用的实验数据量稳步增加。 相比之下,计算机模拟计算方法可以在早期阶段产生大量信息。 然而,信息量的增长与引入体外分析的程度不同。 虽然更深入的计算机分析也可以在后期阶段提供更多信息,特别是通过整合实验数据,但在发现/开发的早期阶段,当有限的或没有实验信息可用时,其主要益处得以实现。(Adapted from Zurdo J. et al. Biomed Res Int, Vol. 2015, Article ID 605 427, 2015. doi:10.1155/2015/605427)
6.1.2 Lead选择
例如,应在实验和计算水平上进行多达10种抗体候选物的lead选择。 引导选择的可开发性驱动方法首先是审查所寻求的QTPP /目标概况并确定关键或限制特征是什么。 通过一系列关键/限制特征,可以选择一组合适的评估技术。 实际上,正如我们在本章前面讨论的那样(第2节),重要的是调整所用的分析方法(信息细节和所需的吞吐量),以及此阶段的资源和材料可用性。 这是使用计算方法的主要动机之一,这在发现和开发的早期可能会有很大的帮助,并且解释需要代理或代理分析(surrogate or proxy analytics)。
除非已经确定蛋白质结构或产生结构同源性模型,否则计算信息的实际量没有改变。但是,由于需要比较的候选物较少,因此可以更详细地讨论。在早期筛选阶段,可以通过代表性评分来估计许多因素,例如免疫原性潜力(参见第4节)。同样,风险评估中可能包含潜在化学不稳定性或降解风险的详细信息。鉴于潜在问题的存在或数量可能已足够,在主要选择/优化阶段,可以考虑,例如,风险类型,其在蛋白质序列结构中的位置,其概率事件,当然,它对活动和安全的潜在影响,作为其关键性的衡量标准(见第5节)。正如我们在上一节中的示例中所示,检测影响最终产品中CQA的潜在化学不稳定性是当今工业中计算方法的最常见用途之一。不幸的是,它们的利用率还没有像人们想象的那样广泛传播,它们的范围仍然有限。
有多种分析技术可用于候选物的生物物理表征,表10.4列出了常用的生物物理方法。然而,正如我们在第3节中讨论的那样,它们对小规模/高通量分析的适用性各不相同,因此了解所提供信息水平与吞吐量,时间和样品要求之间的平衡非常重要。 另一方面,例如,分析超速离心(AUC)是表征蛋白质 - 蛋白质相互作用以及溶液中形成的蛋白质寡聚体的类型和形状的优秀工具,显然信息丰富。然而,尽管近年来取得了进展,但它仍然是一种低通量方法,需要大量的材料和分析时间。因此,AUC不适合于选择最佳候选物或在各种条件下探索配方性或稳定性。这就是为什么选择适合目的的合适工具很重要的原因(参见第3节)。除了候选lead的生物物理特性外,还可以通过引入第4节中定义的临床前免疫原性评估来改善安全风险。 最后,如上所述,可以评估候选相对产量和在给定生产系统上使用的适用性。

在此阶段,理想情况下应使用最终格式和宿主细胞类型比较候选者,即使使用标准缓冲液和溶液。 如果尚未确定最终形式(例如,IgG1和IgG4同种型被认为是合适的支架),重要的是要记住候选物在一种形式中比在另一种形式中表现更好,并且这并不总是可预测的。 虽然在下一阶段讨论了潜在的再造,但如果需要,也可以在此时提供解决特定问题的方法。
6.1.3 lead优化和潜在工程化
lead优化阶段在小分子开发中很常见,但在生物开发中经常被忽视。 尽管有标题,但修改主要候选物的主要序列并不是轻易做到的。 然而,鉴于启动细胞系构建和过程开发所需的资源和监管的跳跃,我们建议在继续之前至少对基于计算方法的候选物进行风险评估。 风险评估应确定是否:
- 流程开发(PD)可以利用平台流程和标准优化来进行
- 通过增强的QbD方法或对一个或多个领域/流程的控制策略,可以降低PD的风险
- 进一步的实验量化和风险验证是必要的
- 应尝试进行蛋白质工程以消除或减少潜在问题
图10.9显示了该方法的示意性工作流程。 计算预测应该在这个阶段变成经过实验验证的信息,并且计算方法的使用从筛选和选择转变为风险评估和蛋白质工程。 即使没有额外实验的余地,计算风险评估也可以突出潜在的可制造性问题,并且可以实施缓解计划或增强的控制策略。 平台流程可能已经通过QbD方法通知,在这种情况下,风险评估和主动管理的控制策略是课程(course)的标准。
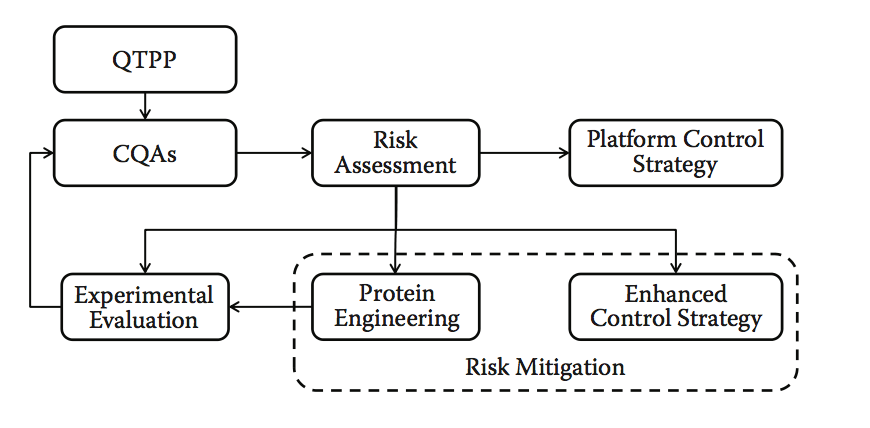
图10.9 预处理开发风险评估和潜在客户优化。 请注意,平台控制策略是指流程开发和制造控制,并且可以反过来包含QbD方法。
最近的两项研究显示了可开发性方法的实际例子,用于评估和降低单克隆抗体的可制造性风险。 在其中一项研究中,设计了不同的缓解策略来解决所确定的问题,包括实施过程控制策略,或者在一个特定情况下,需要蛋白质再造。 这些研究利用分析工具而不是计算作为主要评估标准。 但是,正如我们稍后将要看到的,人们可以设想使用计算机方法可以用于评估给定风险和设计具有改进性能的替代变体的情况。
6.2 实际案例:选择和设计最佳lead candiates
以下部分提供了两个应用前面描述的一些可开发性工具的示例,用于选择或设计具有增强属性的生物制药候选物。 我们选择了两个不同的案例研究来说明它们的实施,以评估或减轻免疫原性或聚集风险。 在这两种情况下,我们都会评估相应的风险,风险缓解和验证步骤的应用,以及计算机和适当的替代或代理体外方法如何在统一的工作流程中结合起来。
6.2.1 案例研究1:选择具有降低免疫原性风险的生物制药:白蛋白结合域
Epibase 电子计算免疫原性预测平台已成功用于许多生物制药中。 这些包括诸如AlbumodTM的工程化,双特异性IgG抗体的去免疫化以及关于抗体工程期间引入的突变对免疫原性的影响的风险评估的表现的实例。
在这种情况下,我们描述了计算机预测工具在选择具有降低的免疫原性潜力的生物制药变体中的应用。治疗性蛋白质的半衰期延长的已知策略是利用血浆中人血清白蛋白(HSA)的长循环半衰期。 Affibody开发的Albumod技术是一种专利的白蛋白结合技术,基于小白蛋白结合域(ABD)。该结构域由5kDa蛋白质组成,该蛋白质经过工程改造以高亲和力结合HSA,旨在通过延长患者的循环半衰期来提高生物制药的功效。 ABD结构域从细菌蛋白质链球菌蛋白G(SpG)中分离,其具有结合血清白蛋白的能力。野生型蛋白ABD001(SpG菌株的白蛋白结合区,ABD3)经历了亲和力成熟,其中一种产生的工程突变体ABD035表现出优异的稳定性,同时对几种物种的血清白蛋白具有增加的亲和力,包括对人血清白蛋白飞摩尔(femtomolar)亲和力。由于已经通过实验证实ABD001含有已知的T细胞表位,因此高亲和力变体ABD035已经通过蛋白质工程进行去免疫。设计了许多变体以除去/减少T细胞和B细胞表位的数量,同时保持热稳定性,溶解度,表达产量和对HSA的亲和力。蛋白质工程阶段由B细胞和T细胞表位预测程序和关于ABD的现有文献指导,并包括迭代轮次的蛋白质表达和分析表征。
随后使用Epibase 计算机平台筛选野生型ABD001和总共133种不同的工程化变体的免疫原性,使用42种HLA II类同种异型对高加索人群进行分析。 基于其结合DRB和DQ同种异型的免疫原性评分对变体进行排序。 然后基于它们的序列,HSA亲和力,热稳定性,溶解度和预测的较低免疫原性风险从变体集合中选择三种变体ABD088,ABD094和ABD095。 图10.10a显示了这些变体的预测免疫原性评分以及它们与亲本ABD001的比较。 与亲本分子ABD001相比,预测免疫原性变体的免疫原性风险降低约40%。
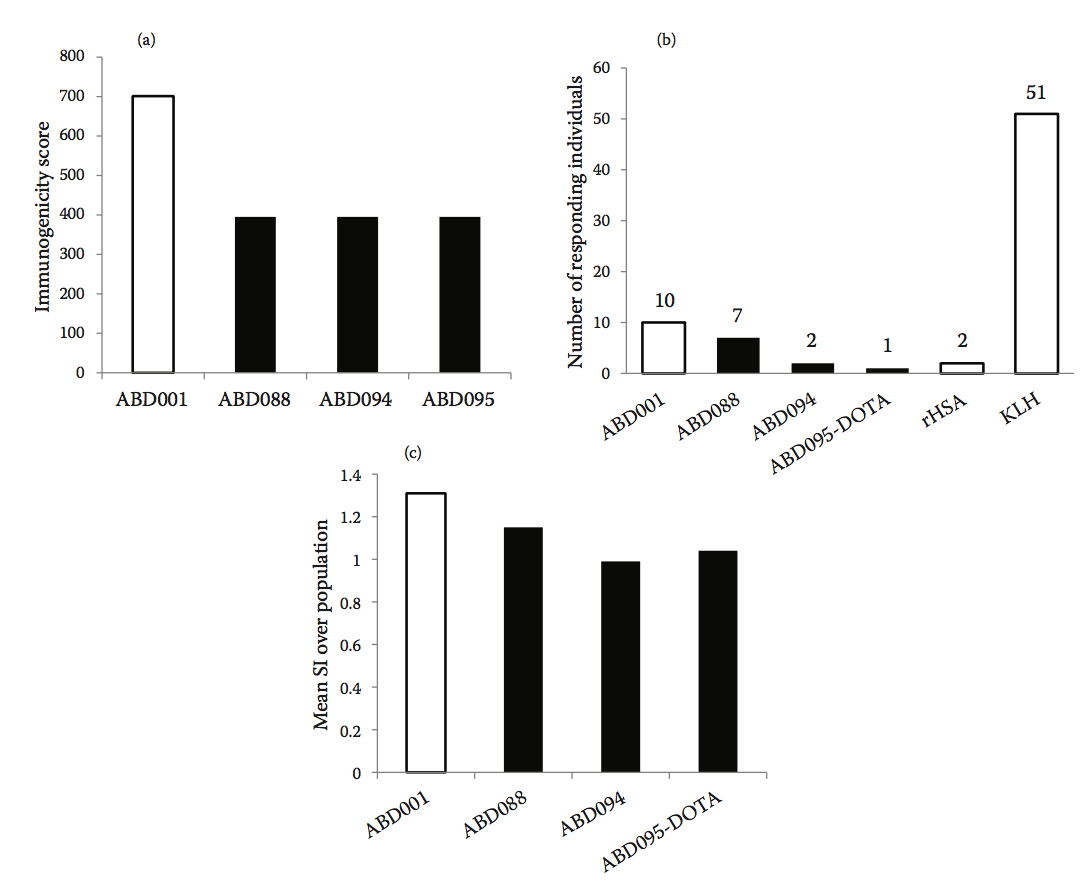
图10.10 (a)预测三种ABD变体和亲本序列ABD001的免疫原性评分。 (b)在52个供体的队列中对ABD变体的CD4 + T细胞增殖反应,突出显示与KLH和rHSA对照相比具有针对ABD变体的增殖反应的供体的数量。 (c)在52个供体的队列中对ABD变异体的CD4 + T细胞增殖反应表示为对群体的平均刺激指数(SI)。 rHSA用作参考(SI = 1)。
在体外进一步评估野生型ABD001和三种去免疫化变体ABD088,ABD094和ABD095-DOTA(DOTA-二价金属离子螯合剂)激活CD4 + T细胞的能力。在体外免疫原性评估期间,CD4 + T细胞的增殖用于监测由ABD变体诱导的T细胞活化反应。在代表高加索人群的52个健康供体的PBMC中评估CD4 + T细胞应答(基于HLA-DRB1同种异型分布的频率)。匙孔血蓝蛋白(Keyhole limpet hemocyanin, KLH)用作高免疫原性基准蛋白,重组人白蛋白(recombinant human albumin,rHSA)作为对照参考。数据分析包括鉴定引发对每种ABD变体的显着CD4 + T细胞应答的个体供体的数量,和对整个52供体群体的平均CD4 + T细胞应答的量度。图10.10b显示了使用空白对照作为参考的具有统计学显着增殖反应的供体的数量。与空白对照相比,52个个体中只有2个对ABD094和ABD095-DOTA有反应,而对应于野生型ABD001的10个捐献者。另一方面,52个捐赠者中有51个回应了KLH阳性对照,只有2个捐献者对rHSA做出了回应。图10.10c显示了使用rHSA作为四种ABD变体的参考的群体的平均刺激指数(SI)。
与野生型ABD001相比,所有三种去免疫化变体均显示T细胞增殖减少。 ABD001和ABD088的平均群体反应与rHSA的平均群体反应(p <.05)(平均SI分别为1.31和1.15)。 ABD094和ABD095-DOTA变体的SI与测试群体相比没有显着差异(平均SI分别为0.99和1.04)。
没有检测到针对先导候选物ABD094的显着的体外CD4 + T细胞应答,表明通过工程去除T细胞表位成功地降低了分子的免疫原性。 作为这些研究的结果,选择候选ABD094进入发育并预期进入临床试验。
该项目证明了成功使用计算机预测和体外免疫原性评估工具的组合作为合适的平台,以指导蛋白质再造去除T细胞表位,并基于不同候选物的相对免疫原性风险实现lead选择。
6.2.2 案例研究2:工程化抗体具有降低的聚集潜力和改进的可制造性
下面我们描述一个例子,用于在简单的工作流程中验证计算和蛋白质工程方法的实现,以解决单克隆抗体中已知的可制造性问题。 该项目描述了通过凝胶渗透高效液相色谱(GP-HPLC)测定的现有亲本抗-IFNɤ人源化抗体的增强抗体变体的产生,其在天然条件下(蛋白质A后)表现出显着的聚集问题。
为人源化抗-IFNɤ抗体的Fv区构建三维结构同源性模型。使用Lonza的Aggresolve计算平台分析分子的序列和结构特性,以评估相对聚集倾向,并识别可证明稳定性和聚集问题的潜在热点或弱区。当与已知行为的参考组比较时,这种分析突出了人源化抗体上的几个潜在的聚集热点。随后,生成针对聚集热点的不同序列变体的文库以及可能影响这些热点的行为的潜在结构liabilities或弱点。该文库通过利用计算机输入和亲本分子的可用信息,丢弃可能对分子的稳定性和结构完整性产生影响或可能影响其生物活性的不适当修饰。在第二次筛选中,选择减少数量的变体用于进一步的体外分析。
使用96孔板中CHOK1SV细胞的小规模瞬时转染,评估所选变体的生产力和聚集。 考虑到他们的生产力和表观聚合,两个变种被列入候选名单以进行进一步评估。 在200ml摇瓶中表达变体,并进行更详细的蛋白质稳定性(聚集)和活性研究。
与亲本人源化抗-IFNα相比,重新设计的抗体显示出显着改善的可开发性。 GP-HPLC分析显示两种选择的候选物几乎完全消除了聚集(图10.11a)。 加速稳定性研究后的相同类型的分析,其中抗体在60℃温育2小时,也显示聚集显着减少,与亲本分子相比,两种变体的单体回收水平增加( 图10.11b)。 此外,与亲本分子相比,重新设计的变体的观察到的表达产量增加了近三倍(图10.11c),与我们组中的早期观察结果一致,将抗体稳定性和聚集与生产力联系起来。 而且,非常重要的是,与亲本人源化序列相比,重新设计的变体也保留或改善了它们的生物活性(图10.11d)。
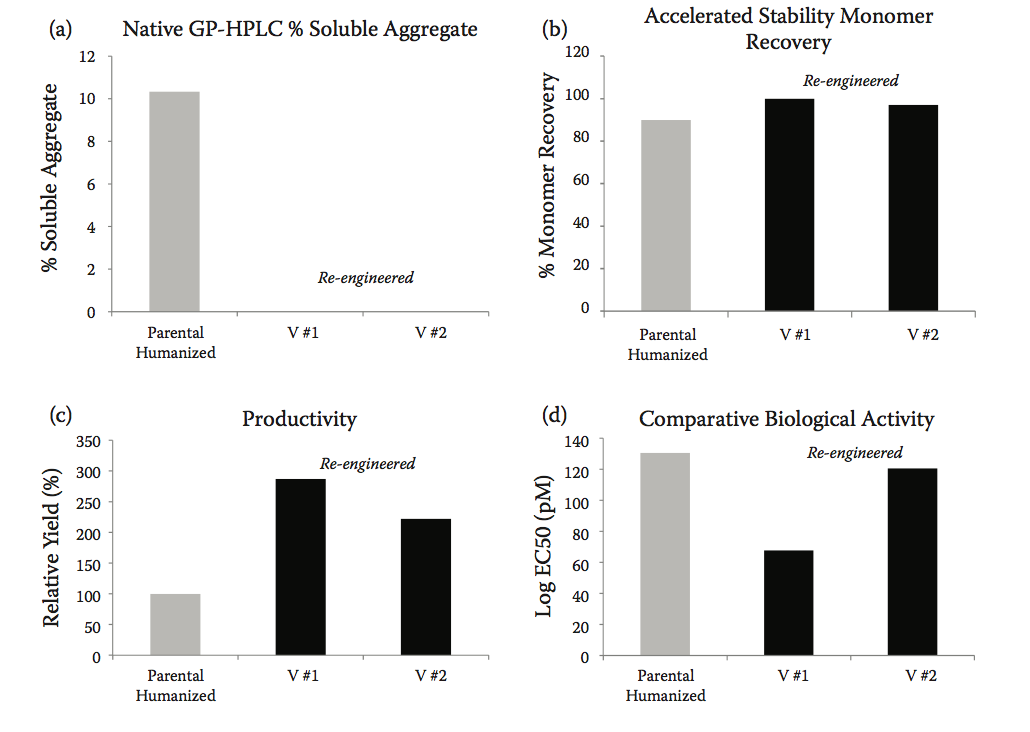
图10.11 重新设计的抗体显示出改善的可开发性。 (a)完全消除了聚集。 (b)在60℃下孵育2小时后的单体回收率在重新设计的变体中增加至几乎100%,表明稳定性提高。 (c)与亲本序列相比,重新设计的变体的表达产量显着增加。 (d)在变体序列中保留或甚至略微改善生物活性(Adapted from Zurdo, J., et al., Biomed Res Int, Vol. 2015, Article ID 605 427, 2015. doi:10.1155/2015/605427.)
人们越来越关注生物制药制剂中亚可见颗粒的存在,因为它们可能对免疫原性风险产生影响。 为解决此问题,使用微流成像(MFI)分析所有变体。 MFI能够基于它们的大小量化给定蛋白质溶液中亚可见颗粒的分布。 在我们的测试中,与亲本分子相比,两种重新设计的抗体变体显示整个光谱中的颗粒显着减少(图10.12)。
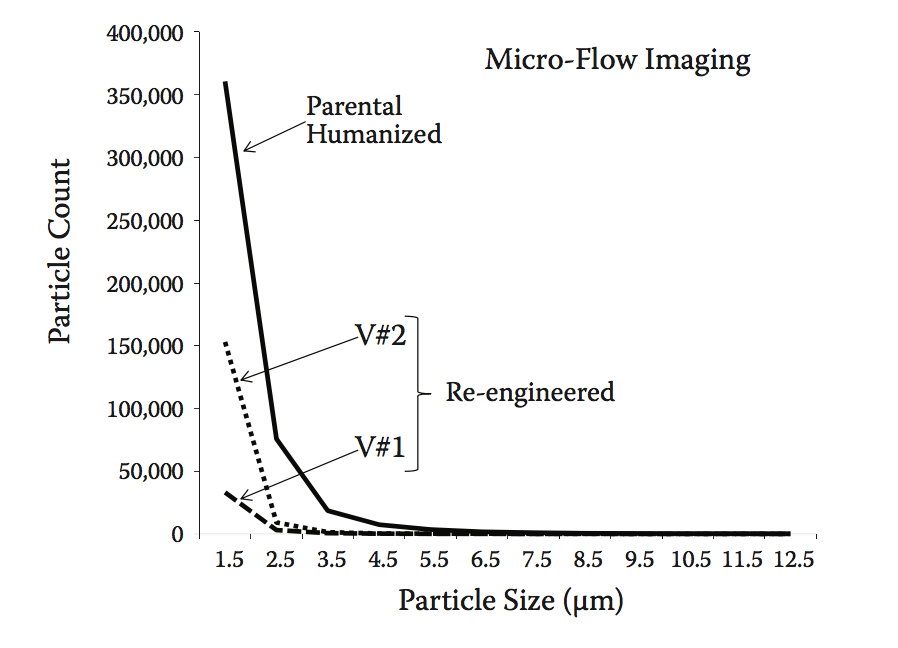
图10.12 通过微流成像(MFI)定义的原始人源化和再造的抗体变体中的亚可见颗粒分布。 与人源化抗-IFNα相比,V#1和V#2变体显示出观察到的颗粒数量减少5至10倍。
这些结果强调了在药物开发的初始阶段,应用正确的计算和分析工具如何能够显着改善给定候选药物的可开发性。 它进一步说明了在重新设计工作中投入的短时间,如何能够显着提高产品的生产率和稳定性,从而降低在临床前和临床开发的后期阶段可能出现的质量和安全问题。
七、实施可开发性工作流程
由于未能完全理解战术前景,已经失去了许多战斗,相应地,由于未能定义和控制终点因素(例如,什么是疾病目标?预期的是什么?或者所需的治疗方式?患者人群的人口统计特征是什么?),更多的前瞻性药物疗法在其管道中出现了某种程度的失败。 在不了解这些特征的影响的情况下,治疗可以一直进行到最终用户,然后不能按预期对患者群体进行。 正是出于这个原因,我们提请注意将这些参数纳入预期性能概况(QTPP)的关键性,而不是限制系统和流程,以仅解决与生产或测试直接相关的属性,而是采取更全面的方法 风险管理。
QbD方法的实施要求定义质量目标产品概况(quality target product pro le,QTPP)作为绩效的基础,并确定那些关键的质量属性(CQA),并且需要谨慎控制以保持产品的完整性和功效。如前所述,ICH Q8及后续指南的实施主要集中在QbD的制造过程方面,这往往导致将目标产品概况(TPP)的定义限制为参考产品中存在的方面,以及由不同分析方法定义的特定特征。在这里,我们论证了实施工作流程的情况,该工作流程将可开发性方法纳入其核心。图10.13说明了如何构建这样的工作流程。该工作流程要求在药物开发过程开始时正确定义QTPP,理想情况是在早期发现阶段(产品设计),以便尽可能详细地制定预期的性能,安全性和经济目标概况,最终确定药物开发过程中的最终产品目标。从该起点开始,可以导出CQA并且实施合适的可开发性风险评估,以导出匹配所需CQA的最佳候选者或重新设计先导候选者以适应目标概况。实际上,在设计阶段,应该在主要候选物中绘制和引入或选择这些属性,但当然,它们也应该是制造过程的设计和优化的一部分,以便可以适当地控制这些属性。一种有效的方式。人们可能会期望在候选物设计和选择过程中早期引入的降低风险的方法,将反过来增加制造过程的稳健性,从而更容易控制特定的CQA并最大限度地减少偏差或超出规范的发生率( OOS)发生。
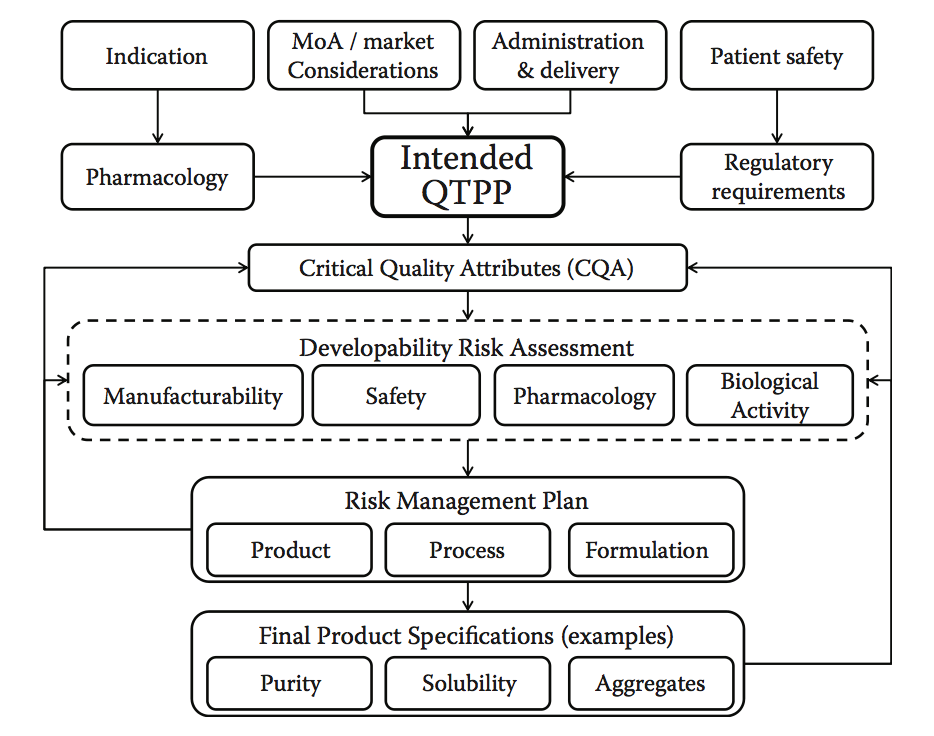
图10.13 可开发性流程图。 该流程图说明了如何明确制定发展计划,以有效整合可开发性风险评估工具。 基于指示,药理学,作用模式,市场和交付等设置预期的性能概况(QTPP),允许开发者确定应该评估主要候选者和过程的CQA。 可开发性风险评估有助于识别影响这些CQA的特定风险,并设计和实施风险缓解计划。 这可能涉及对主要候选物的选择或设计进行修改,潜在的再造(产品),旨在最小化或控制风险的制造过程的特定要素的设计,或者可能涉及某些特定的配方要求。 所有这些步骤将根据可测量和可控特性来定义最终产品规格。(Adapted from Zurdo, J., et al., Biomed Res Int, Vol. 2015, Article ID 605 427, 2015. doi:10.1155/2015/605427.)
构建发展计划的过程围绕一系列不同的阶段,这些阶段反过来解决以下四个问题:
- 什么样的质量特征需要纳入药物和药品特征? (定义QTPP)
- 哪些特征和属性构成风险并被视为关注的领域,为什么? (定义CQA)
- 哪些属性对治疗分子的理想性能至关重要? (可开发性评估,关键性)
- 在发现和开发阶段需要引入哪些缓解计划? (补救计划)
7.1 STARTING WITH THE END IN MIND :QTPP和CQAS
为了说明整个过程,我们设想了一种虚构的生物制药产品,以帮助提出一些关于顶级特征的背景,这些特征应该被用来定义QTPP和合适的工作流程。我们在此强调,QTPP的定义不是一项简单的任务。它需要来自多个学科和发展领域(发现,制造,临床开发),供应链,分销等的技术专家的参与。最重要的是,它还应该包含最终用户的关键输入需求和要求,或者QbD命名法中被称为客户的声音。值得注意的是,最终用户或利益相关者应在广义上进行定义,以确保成功(与上文讨论的Juran的“大Q”定义一致),并应包括患者,临床医生,付款人,医疗保健提供机构等等。 QTPP将最终定义给定产品的要求,因此可以认为它适合于目的。这些可以分为三类,与前面提到的三大支柱一致:
- 适合指示(indication)。 适用于所需的疾病状况,给药方案,患者群体,给药途径和所需的PK / PD。 (作用方式和药理学)
- 适合过程。 可以使用标准工艺以所需规模制造。 它足够强大,可以承受过程偏移而不会显着影响CQA。 它足够稳定,可以满足工艺和配方要求。(制造性)
- 适合患者(安全)。 在不影响患者安全的情况下实现期望的治疗结果。 不会引入潜在的危险副作用。 (安全)
7.1.1 想象中的生物制药
为了说明这个过程,我们选择了一个想象中的生物制药产品和一些需要满足的虚构属性和要求,因此我们可以提出一个合适的开发工作流程,其中包含可开发性方法。 让我们考虑A公司开发针对自身免疫疾病的创新疗法。 该公司已经确定了两种新的药物靶点(A和B),这些靶点有可能开发针对类风湿性关节炎(RA)患者的难治性(即对现有治疗无效)的新疗法。 已经使用Fab片段抗体展示文库平台为两个靶标中的每一个选择了潜在结合物的集合。
基于在动物研究中获得的有希望的数据,该公司打算为每个靶标A和B选择合适的结合Fab片段,并将它们组合成双特异性抗体形式,具有中和/阻断作用模式。该公司正在为最终设计探索不同的多特定平台。由于使用常规抗体下游平台可延长半衰期和制造准备,因此含Fc结构是有利的。该计划最初定位为难治性RA的治疗,但也应该能够在可负担性和患者依从性方面保持现有治疗的竞争性替代方案,并且可以在家中使用的笔式输送装置促进。该开发公司最近完成了一系列B轮融资,其中包括一些风险投资基金,用于完成其两个项目的概念验证研究(第二阶段临床试验),包括此处提到的RA项目,并打算与合作伙伴合作其中一家拥有一家大型制药公司或生物技术公司。投资在许多不同的支付中交错,并且是里程碑式的。
在这种特定背景下,许多潜在的挑战可能会影响A公司提出的发展计划。通过定义适当的QTPP来指导针对该产品定制的发展计划,可以充分探索这些挑战。 这样的发展计划将纳入适当的风险缓解要素,以直接解决或至少改善QTPP强调的这些挑战所带来的风险。
7.1.2 定义合适的QTPP
正如我们之前所说,治疗产品的QTPP定义不是一件小事,需要来自多个利益相关者和学科的意见。在这里,我们只提供一个列表,其中包含一些可能与为我们的虚构产品定义QTPP相关的特征。其相关性被强调为潜在的关注领域或要求。请注意,要求不仅与制造方面有关,而且主要与最终用户的要求和特征有关,例如产品的使用方式和目标患者人数等。简而言之,QTPP特征将涉及期望的适应症,患者群体,药物靶标和给药方案,递送途径和方法,目标指示和市场,制造平台,要使用的特定分子形式和产品的固有性质。表10.5汇编了有助于定义QTPP的特征和潜在需求列表。

重要的是,QTPP应考虑药物的整个生命周期,从设计和制造到分销和患者管理,甚至非常重要的是,它可能在未来的其他适应症中使用(例如,儿科克罗恩病) 可以包含对产品非常不同的特定要求。 在这种特殊情况下,A公司的商业模式也会带来额外的限制因素。 特别是在这种情况下,还应特别注意未来设想的合作伙伴(即大型制药公司或生物技术公司)在格式,制造平台,质量和监管方面,行动方式等方面的潜在要求。 实际上,这可能会损害商业化尝试和未来的投资回合。
7.1.3 推导CQA
通过定义涵盖QTPP定义的所有领域的产品的预期性能概况,可以推导出相关的CQA,以在整个药物开发过程中设计适当的风险评估和控制管理计划。 从控制或风险缓解的角度来看,CQA可分为三大类:
- 可通过产品设计控制(即半衰期,T细胞表位)
- 可通过制造工艺控制(即糖型分布)
- 可通过产品设计和制造相结合进行控制过程(即稳定性,降解)
表10.6总结了基本CQA和受影响的QTPP区域的列表,以及潜在的控制方法。我们使用ICH Q6B和ICH Q8(R2)作为定义CQA的基础,并进行了一些额外的修改。例如,请注意我们将免疫原性作为CQA包括在内。 CQA通常限于可通过特定分析方法评估的药物或药物产品的那些方面或特征。可以容易地测量和表征聚集,稳定性,杂质,甚至生物活性(通过适当的生物活性代用测定,例如与固定的靶蛋白结合)。然而,正如我们之前讨论的那样,生物制药产品的安全性问题,如免疫原性,是一个不太明显的方面,传统上只能在临床环境中妥善解决。然而,我们相信,新技术和分析(例如本章和本书其他部分所述的那些)的出现现在可以提供用于描述其他产品属性及其相对风险的合适平台。此外,为了进行早期风险评估,我们已经使用本章前面和本书其他地方描述的方法将CQA的数量限制在可以在后期发现/早期开发中使用的区域。

在我们特定的虚构例子中,目标适应症(自身免疫性疾病),所需的给药方案(慢性治疗)和选择的给药途径(皮下)都是已知的免疫原性因素,其产品潜在的免疫调节作用可能进一步加剧。需要在早期解决所需的药理学性质,例如半衰期,因为它们可能影响预期的给药方案,给药途径,制剂等。在这种情况下,公司A选择的解决方案是使用含有Fc部分的分子形式,其将对半衰期产生积极影响,并促进下游加工。这是一个很好的例子,说明如何通过直接在产品中设计CQA来轻松解决这些问题。此外,管理的途径和形式很可能对产品的配方,长期稳定性和储存产生强烈的限制。许多CQA,例如溶液粘度,产品溶解度,稳定性(热,光敏性)和聚集,需要在设计阶段早期适当地解决,因为它们可能影响许多不同的QTPP区域。
从制造角度来看,纯度和产量也是需要考虑的重要因素。 这些应该满足目标指示的预期市场需求并且适合合理的制造能力。 这意味着细胞系的生产力特征可能是重要的,特别是如果选择的产物(双特异性抗体)具有低生产率/过程产量的迹象或者包含异常复杂的下游过程要求。
7.2 可开发性评估:定义风险区域和减缓战略
按照图10.13所示的工作,一旦定义了CQA,就会产生可开发性风险评估。 在表10.7中,我们展示了一些可以通过实施本章所述的不同可开发性方法来处理的潜在风险领域的示例。 此外,我们已经展示了如何通过评估发生概率(计算机预测分数或体外分析)以及给定风险的潜在影响来定义相对风险(临界)。 在此,我们根据之前定义的QTPP要求,假设所描述的某些风险可能对产品的可行性/成功产生影响。
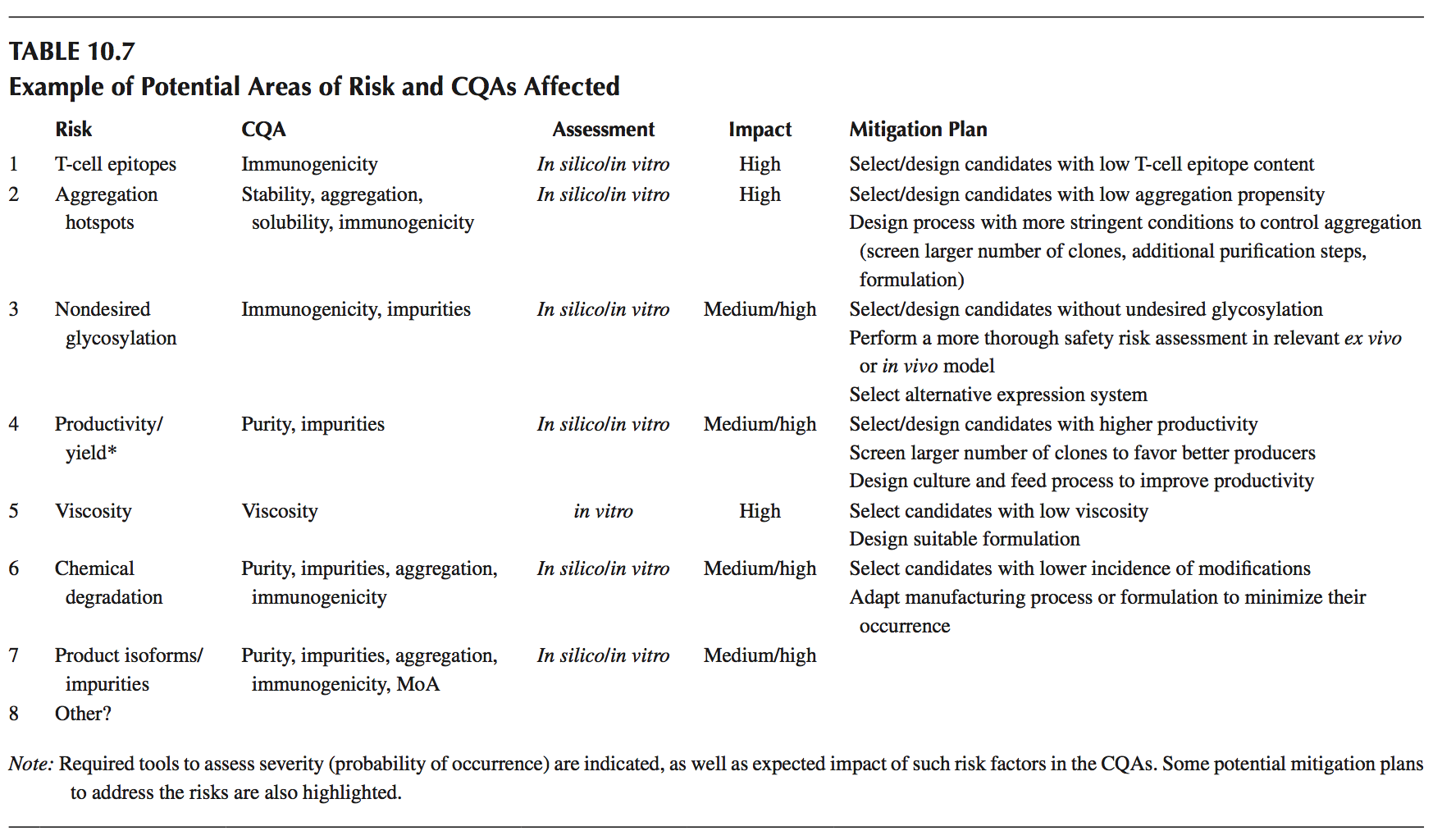
由于缺乏对这种特定结构(多特异性抗体)的经验,我们已将生产率/产量作为评估的重要风险。 这是基于这样的理解:与标准抗体分子相比,多特异性构建体通常存在不同种类的制造困难。
鉴于该产品可能是通过将两种不同的特异性(来自两个独立的Fab展示文库)组合成单个分子而设计的,因此有可能在将两个构成的Fab片段组合成单个片段最终分子之前分别解决可能的CQA问题。例如,可以根据它们在若干领域中的行为来选择/设计单个单特异性Fab,例如聚集潜力; 稳定性,溶解度和粘度; 潜在的PTM(不希望的糖基化); T细胞表位; 和相对生产力。
从所有已识别的CQA的集合中,可以将焦点缩小到与分子的期望性能特别相关的那些风险。 使用本章前面部分概述的策略,可以通过计算机预测方法或合适的代理分析或体外测试评估其发生概率来解决这些潜在风险。 关键性评估(定义较早)将结合这种发生概率及其在产品QTPP中的相应影响。 如果给定的特征被认为是关键的,那么可以引入适当的缓解计划。
例如,计算机分析可以鉴定Fab片段之一的结合区域中潜在的降解发生。这可能意味着抗原结合的丧失以及产物不稳定性,聚集和免疫原性风险的增加;因此,这种风险可被视为具有高影响力,由此产生的关键性评估将建议实施缓解计划的必要性。一种潜在的缓解策略可以是将分子推进到可配方性评估中,以便确定可以最小化其发生的pH范围,溶液条件和赋形剂。或者,蛋白质工程可以潜在地消除降解风险,希望不改变生物活性或其他基本产品特征。在某些情况下,蛋白质工程和配方性评估的双向方法可能是可取的,特别是在其他所需的CQA包括高溶解度或低粘度(特别是在高浓度下)的情况下。
如第10.5节所述,可以通过根据不同风险在所需QTPP中的相对影响,来定义不同风险的相对权重来实施综合评估。 这可以在很短的时间内促进许多不同候选物(即,通过展示方法鉴定的Fab片段)的评估,并且基于与QTPP相关的特征的组合来集中选择最佳候选物。
7.3 设计合适的开发过程工作流程
最终,有许多限制因素将决定如何阐明可开发性工作流程和开发计划。 例如,潜在候选物数量,材料可用性,所用分析平台等元素可以在何时以及如何评估不同风险时发生显着变化。 在理想的世界中,从一开始就拥有关于所有相关CQA的相关信息,将使得有可能找到不同属性的最佳组合来选择/设计领先候选者以在开发中向前发展。 那一天还没有到来,但在本章中我们提出了许多可用于拓宽生物治疗选择标准范围的方法。
无论如何,定义包含适当风险缓解要素的发展计划非常重要。 这将包括:
- 明确定义所有必要的质量属性,这些属性需要作为QTPP的一部分纳入产品中。如前所述,这应该是在创建产品概念以最大化成功概率时。
- 确定哪些部门将参与实施不同的风险评估和缓解策略。这可能需要跨发现和开发功能的多学科方法,需要正确定义。
- 就风险缓解方法达成一致,包括可实施的可开发性工具以及可在何时何地引入,以及利用风险影响评估和决策方法。
- 就产品及其开发阶段可接受或足够的风险缓解方法类型达成一致。例如,在某些情况下可能需要工程策略,而在其他情况下可能建议采用早期工艺设计和优化。
- 定义路线图,确定在开发过程中需要解决的一些CQA以及应如何解决这些问题,在时间,资源,投资以及可能需要的其他开发或支持技术方面做出足够的补贴。
八、结论和未来前景
有人质疑开发新药的传统模式是否足以满足医疗保健提供和患者不断变化的需求以及更严格的监管要求。事实上,与其他行业相比,药物开发是一个特别漫长,成本高且效率极低的过程,可能带来严重的财务和社会后果。新药的开发遵循根据特定学科或发展阶段细分的高度严格和分层的过程。因此,这种结构在出现(经常)问题时会特别强调解决问题,而不是先于先发制人或先前采取纠正措施。在药物设计和开发的早期阶段,不同发展领域之间缺乏整合尤为明显,其中职能通常彼此完全隔离。这些被分成功能孤岛的结构,实际上是引入行业新实践的重大障碍,例如转化医学的实施,设计的质量,或投资组合生命周期管理。
可开发性最终是一种结构化方法,可将更广泛,更全面的质量解释引入生物制药开发。 因此,它成为扩展QbD在药物开发,和特别是生物治疗药物开发中目前仍然有限的实施的重要工具。
关于QbD在药物开发中应用的现有ICH指南(Q8-Q11),主要涉及为过程理解和表征提供框架。 但是,我们认为真正的QbD始于产品本身的设计。 在本章中,我们试图通过在药物开发开始时尽早定义有意义的质量目标产品概况(QTPP),来说明如何轻松部署有意义的CQA和有效的风险管理策略。 只有在开始时具有这些明确的设计要求时,才能更有效地开发适当的制造工艺并且具有更高的成功概率。
这在生物制药药物开发的情况下尤其重要,其中许多重要性质在设计阶段未得到适当控制并且在制造过程本身期间待确定。实际上,制造过程可以理想地用于控制产品中的特定质量属性,但是这在很多情况下以高成本大大增加了风险,和可能需要迟早解决的失败的可能性。在本章中,我们建议一个潜在的工作流程,从经典的线性层次发展模型转变为更加整合的模式,并且适当的早期风险评估工具可以在很早的阶段帮助控制CQA。在这个工作流程中,重点发生了变化,从一开始就定义了QTPP,并且有大量标准最终定义了给定产品的成功(行动模式[MoA],目标患者人群,交付要求等 )。其次,它涉及引入额外的降低风险的工具,以提高候选物选择的严格性,以便从开发开始就满足所需的QTPP并适当地控制产品中的CQA。
此外,我们认为引入此类早期风险评估范例不仅可以带来经济效益并降低开发成本,还可以加速新产品候选产品的开发,例如加快从临床前到临床开发的转变,以及 最终为患者提供更有效和负担得起的药物。
8.1 行业趋势和差距
在过去几年中,关于生物治疗药物,其作用机制以及制造过程的实质性改进的科学知识不断增加,随着“组学”(基因组学,转录组学,代谢组学)和新基因编辑技术的应用,它们正在经历新的复苏,以根据特定的工艺和产品要求定制个体的特性。 这些包括营养缺陷型敲除的产生,有利于严格的选择条件和更高的生产率,或操纵产物所呈现的糖基化模式。 同样,对代谢需求和new media and feeds 的更好理解使生产力成倍增加到20年前难以想象的水平。
鉴于这种背景,越来越需要扩展生物制药产品设计和选择中使用的标准,以尽可能避免可能危及特定药物发现计划可行性的潜在问题。正如我们在本章中所讨论的,越来越多地使用计算工具来模拟产品性能可能是一种强大的方法,可将QbD的产品和流程知识方面整合到一个无缝结构中,在这个结构中,发现和开发一种成功的治疗产品被视为不可或缺的互连部分。尽管如此,它们的实施还处于起步阶段。需要新的和更好的计算工具,特别是围绕产品和过程之间的交互建模,以便更好地理解和设计合适的制造过程。此外,需要增加这些类型的计算方法的验证工作,以便促进它们作为整体的行业的接受和实施。还需要新的方法来对产品质量属性进行早期分析评估,从而允许更高的产量,更短的时间,并且需要更少量的产品。例如,早期的可配方性评估正在成为一个越来越受关注的领域,但其成功将与开发适应性分析和计算方法紧密相关,这将有助于其早期应用,甚至可能在最终候选物被选中用于制造业发展之前。
8.2 对可开发性的障碍
实施这种早期风险管理愿景,并实现可开发性方法所承诺的真正潜力存在许多障碍。也许对于许多人来说,这个概念可能过于模糊,无法在行业中流行的药物开发结构中实施。正如我们前面提到的,发展过程的分散是一个关键障碍。在这种情况下特别重要的是生物制药药物的发现和开发功能之间仍存在巨大差距。这种分裂存在历史和技术原因,但本章和本书其他地方描述的新技术和方法学进展正在以几年前难以想象的方式将这两个不同的宇宙结合在一起,创造出可以互动的新界面并解决常见问题。另一个重要障碍是采用创新的地方性保守,有时基于监管问题,成本或无法适应现有流程或基础设施而证明其合理性。这在生物制药制造业中是显而易见的,在这种制造中,小的变化会对产品质量属性产生破坏性影响,伴随的风险可能会从批次失败到产品召回等方面发挥作用。实际上,生物制药生产面临着不成比例的压力,无法为特定产品提供所需的质量,这可能是因为在设计阶段,人们很少意识到后期可能出现的困难和并发症。
在这里,可开发性成为桥梁,很早就提高了CQA意识。 但它也可以帮助整合发现和开发活动,并使他们的目标与开发更好,更安全的产品相一致(图10.14)。 幸运的是,正如本书所反映的那样,行业中有一些成功的例子表明这两个功能之间的集成程度不断提高。
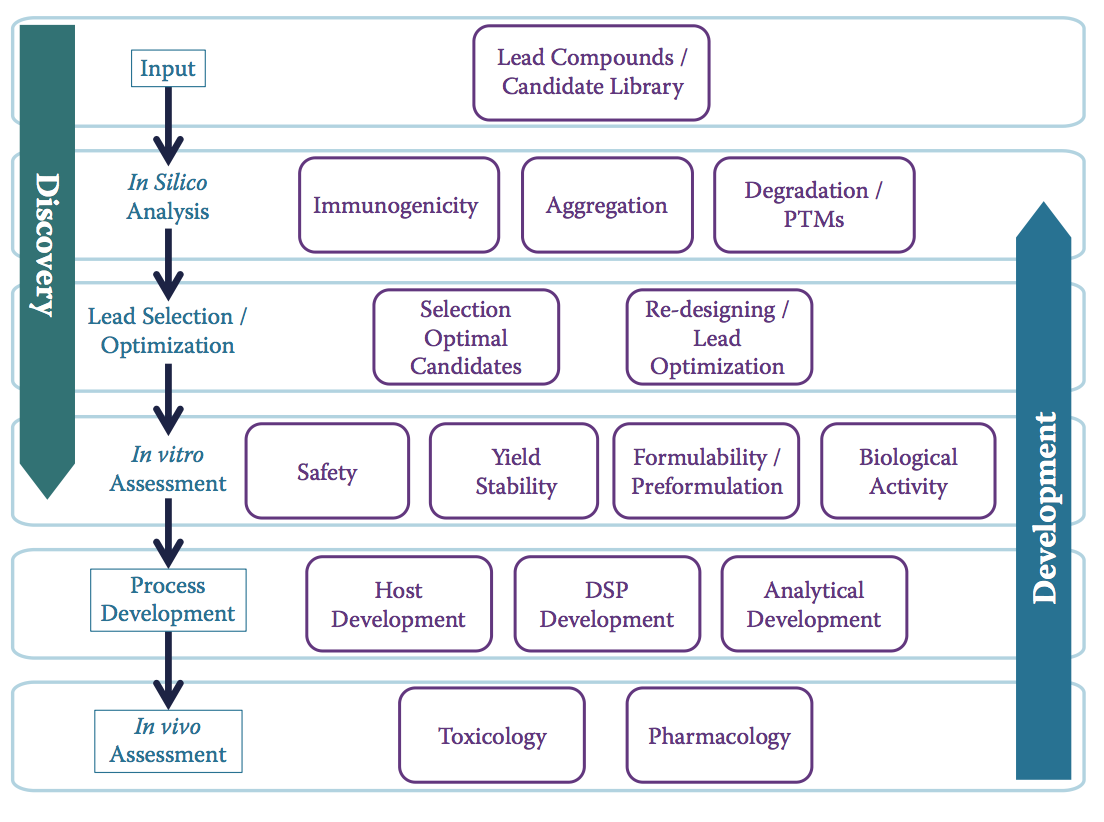
图10.14 作为桥接工具的可开发性。 该示意图显示了重叠的评估层如何融入经典责任区之间,带来了许多好处:(1)促进各职能之间的互动以及发现和发展阶段之间候选物的过渡,(2)促进早期和廉价消除 有问题的候选物,以及(3)帮助设计所需的特征,这些特征可以减少后期开发中的失败,无论是制造还是临床。 突出显示的活动(盒装)对应于传统生物制药开发的标准阶段。(Adapted from Zurdo, J., Pharm Bioprocess 1: 29–50, 2013.)
8.3 展望未来:可开发性,QBD和产品生命周期管理
设计和开发之间的这种集成将需要增加对更复杂的计算模型的利用,并扩展我们关于产品和过程甚至产品和患者之间的相互作用的知识。 这将有助于更好地了解宿主生物学以及如何控制它并设计它以满足特定的制造和产品要求。 此外,在其生物背景下对产品行为进行建模的能力可以帮助识别今天只能在临床中发现的潜在问题。 在其他领域,模拟蛋白质 - 蛋白质分子间相互作用以及蛋白质 - 界面相互作用将为更先进和精细的工具打开大门,以选择和设计更有效的制造工艺和配方。
最重要的是,能够整合科学投入和统计数据分析的强大决策工具的开发,将简化候选物在不同发展阶段的进展,并提供其他行业已有的透明度和可见度。 产品生命周期管理(PLM)是如何进行整合的一个例子,允许采用整体的药物开发方法,从概念阶段到临床评估,考虑到药物产品生命周期中影响药物产品的所有不同方面, 分配,退休(retirement)或更换。 有一些有趣的例子说明如何实现这一点,最终与我们在此描述的QbD综合方法保持一致
然而,这不是一件小事。 可能需要某种协议或定义良好实践,涉及行业中的不同利益相关者,以便为实施可开发性方法定义合适的工作流程。 理想情况下,这将包括监管输入以及与现有QbD指南的整合,例如,通过扩展ICH Q8(R2)的范围,以纳入本书中介绍的可开发风险管理方法。 幸运的是,这方面有一些动作。 其中一个例子是监管机构鼓励探索新的临床前免疫原性评估方案作为临床免疫原性风险的预测因素.
致谢
我们感谢 Affibody AB,特别是Caroline Ekblad,LarsAbrahmsén和IngmarieHöidén-Guthenberg,感谢我们分享本章中包含的一些数据并提供宝贵的意见。 我们还要感谢Rebecca Michael参与本章所涉及的一些工作,以及Yvette Stallwood,Rajesh Beri,Anne Moschella和Hilary Metcalfe,以及许多客户和合作者,讨论这里包含的主题时,他们提供了非常宝贵的意见
参考资料
- 《Developability of Biotherapeutics》
