【5】机械药代动力学-药效学模型在生物制剂开发中的应用(Application of Mechanistic Pharmacokinetic–Pharmacodynamic Modeling toward the Development of Biologics)
一、介绍
自1982年人类胰岛素首次被美国食品和药物管理局(FDA)批准以来,蛋白质和肽作为治疗剂越来越受欢迎。 使用蛋白质作为治疗剂是一种明显的治疗方法,因为许多疾病是由蛋白质突变(如β-葡萄糖脑苷脂酶,乳糖酶,胰酶)或过量或缺乏(如胰岛素,生长激素,血液凝固因子)引起的。 迄今为止,已有超过130种蛋白质或多肽被FDA批准作为治疗药物,超过350种单克隆抗体(一类蛋白质治疗剂)处于临床前和临床开发的不同阶段。
本章重点介绍PK / PD和系统药理学模型在解决生物药物发现和开发关键阶段的关键问题中的应用(图5.1)。在早期阶段,当一个目标被探索作为一个新的干预点时,系统药理学模型可用于检查目标在疾病病理学中的作用,并检查该方法的可行性,因为生物途径无处不在。在靶标验证后,鉴定能够以高选择性和效力抑制靶标的先导化合物变得至关重要。在这个阶段,可以应用包含体内靶标表达和周转率信息的机制模型,以提供关于先导生物制剂的亲和力要求的指导。我们提供了关于针对常见类型的受体 - 可溶性和膜结合的亲和力要求的一般指导。一旦选择具有理想亲和力的先导化合物用于临床开发,可以应用经验PK和PK / PD模型来预测感兴趣组织中的人PK和靶标覆盖。在最后几节中,我们介绍了具有非线性PK的生物制剂的TMDD模型的基本概念,以及针对驻留在特定组织中的靶标的生物制剂的SoA模型。这些模型有助于设计FIH试验并确保机制的临床试验。
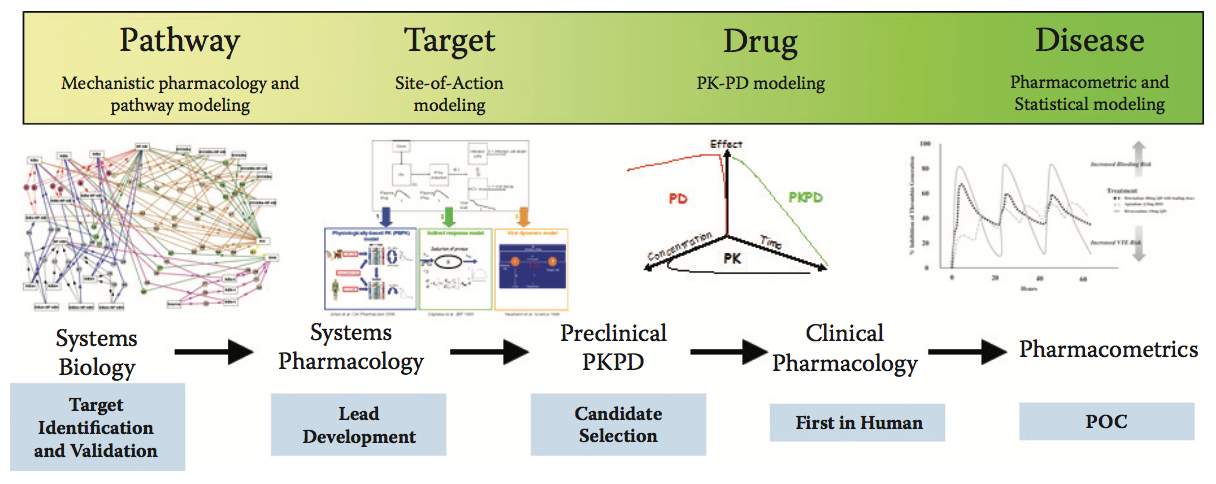
图5.1 在整个药物发现过程中使用建模和模拟方法。
以下部分重点介绍一类称为单克隆抗体(mAbs)的蛋白质治疗药物,其中40种已获得FDA批准。 这些mAb通过向细胞递送有效负载( delivering a payload )或隔离目标靶标( 受体或配体 )起作用,从而干扰引起疾病的机制。 本章的各个部分重点介绍了用于解决药物发现各个阶段出现的特定问题和问题的关键建模和模拟(M&S)方法(图5.1)。
1.1 单克隆抗体同型 MONOCLONAL ANTIBODY ISOTYPES
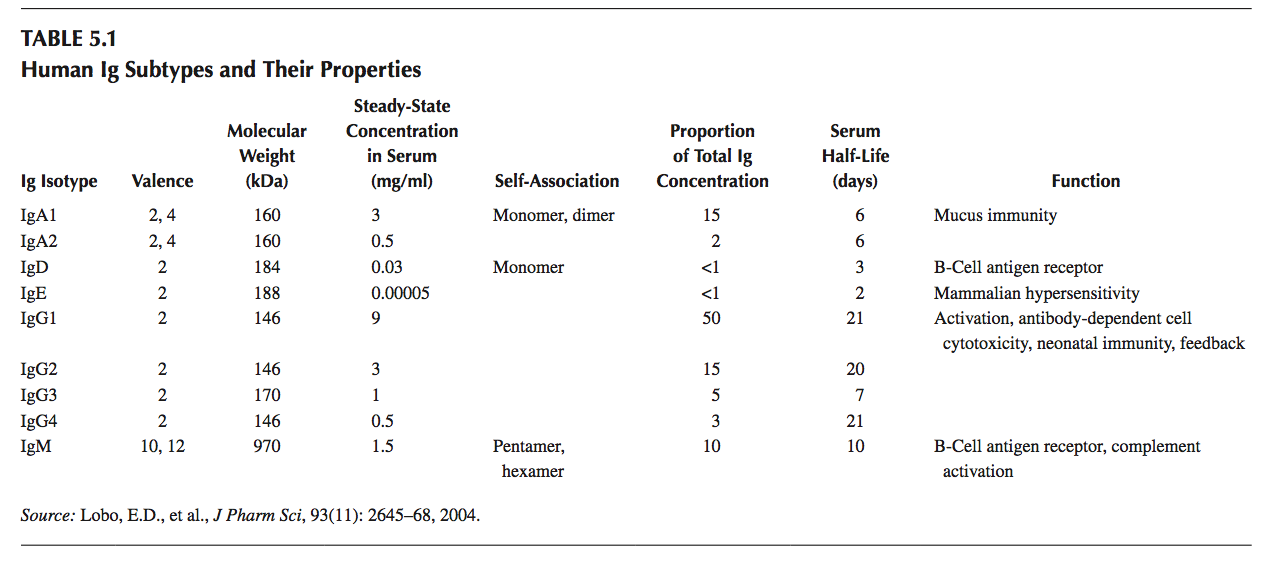
mAb是一类属于免疫球蛋白Ig超家族的分子,含有两条相同的重链和两条相同的轻链。 根据存在的重链的特征,mAb可以进一步分为5种不同的同种型(isotypes)(即IgA,IgD,IgE,IgG和IgM),如表5.1所示。 人体中存在两种轻链同种型:κ和λ。 重链和轻链均含有识别其靶标上的表位(epitopes)的互补决定区(CDR),例如可溶性蛋白质或细胞表面受体。
1.2 特异性和亲和力
mAb设计有表位,使得它们对靶标具有高度特异性和亲和力。 这种组合使它们具有靶向抗原的能力,同时减少对周围组织的任何不良影响。 第3节详细讨论了亲和力对mAb与其靶点相互作用的影响。
1.3 免疫原性
有许多因素可以使分子具有免疫原性。 如果治疗剂的Ig来源是基于动物的(小鼠或部分小鼠),则几乎可以肯定免疫原性应答的可能性(图5.2)。 由外来蛋白质引起的这种反应可能导致一系列可能严重过敏的事件,从而导致灾难性后果。
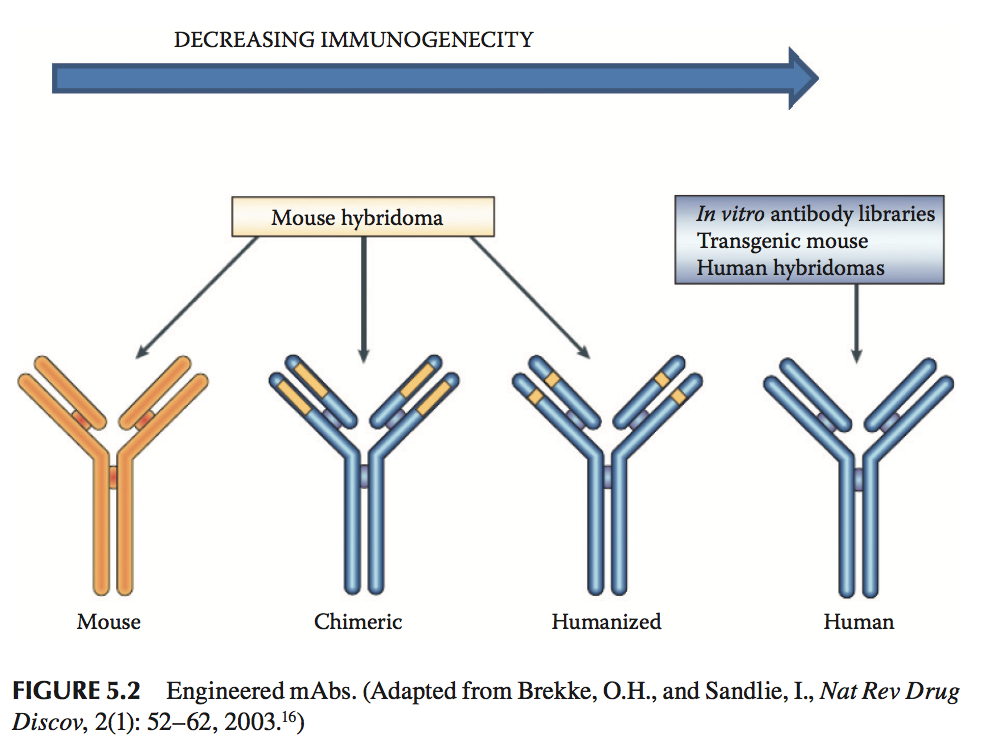
相对良性的作用是产生针对治疗性抗体的中和抗体(neutralizing antibodies)。 这些中和抗体与治疗性抗体结合并有效地将它们从循环中去除,使得治疗无效。 不幸的是,由于适应性免疫系统的“记忆”,这种中和反应可能成为治疗的终身问题。 其他细节超出了本章的范围;
尚未确定控制免疫原性的机制,但已提出若干患者,治疗和产品相关的风险因素。 已经使用数学建模研究了这些风险因素中的一些以获得对免疫原性背后的复杂免疫应答的定量理解。 最近,开发了由亚细胞,细胞和全身模块组成的系统级模型,其概括了针对治疗性蛋白质产物的T细胞依赖性免疫原性的关键过程。 后来该模型还用于评估治疗性蛋白质产物的聚集体是否可以诱导T细胞依赖性免疫应答。
1.4 单克隆抗体药代动力学
mAb的分子大小决定了它与人体的配置(disposition),并且由于大多数mAb是IgG分子,它们的处置行为(disposition behavior)与内源性免疫球蛋白非常相似。 如表5.1所示,IgG具有最长的半衰期(~21天),并且是治疗开发中最常用的构建体。
如果剂量导致血浆水平比目标浓度高至少10倍,则静脉内(IV)施用IgG治疗性抗体表现出典型的双指数衰减过程(这种情况的原因将在3节中阐述清楚)。通常,分布主导的相(α相)持续约3-7天,然后是20-27天消除主导的相(β相)。假定这种典型项目的药代动力学(PK)参数在药物发现的早期阶段(前期开发)期间开发的模型中是已知的。重要的是要注意,这些参数仅在mAb的PK为线性时才有用;也就是说,预期的mAb浓度明显高于目标浓度(≥10倍),特别是如果目标是膜结合的话。 IV或皮下(SC)剂量的典型PK参数总结在表5.2中,并且在没有附加信息的情况下,这些用作模拟中的第一近似值。

1.5 单克隆抗体ADME 性质
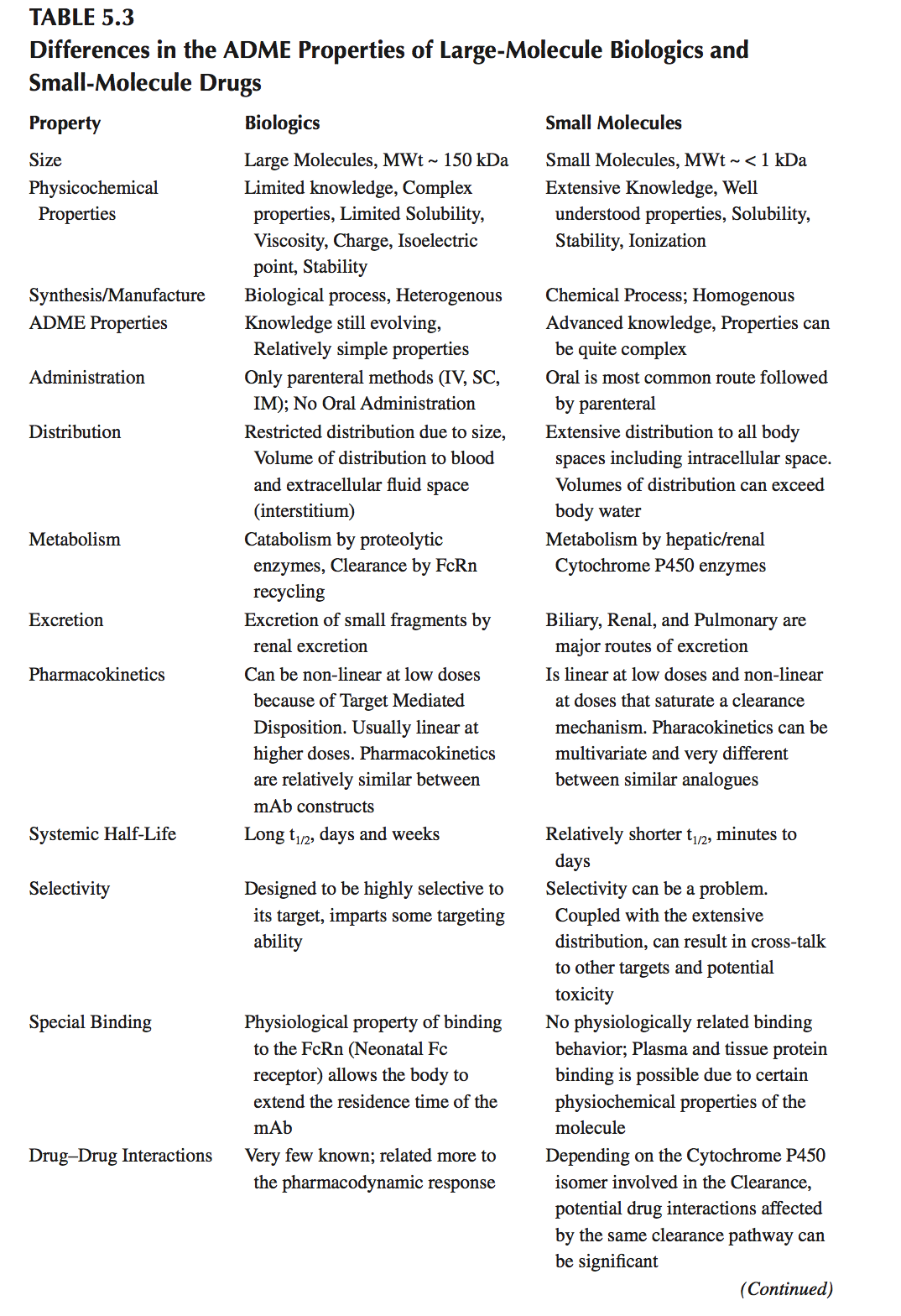
本节重点介绍单克隆抗体的基本ADME特性; 为了更一般的理解,读者可以参考几个关于这一主题的综述。大分子生物制剂和小分子药物的ADME特性的差异见表5.3。

1.5.1 吸收 Absorption
大多数mAb是肠胃外给药(administered parenterally)的,因为由于前面所述的原因,通过肠道途径的吸收受到限制。虽然IV给药很容易,但需要医学专业人员来管理。另一方面,SC和肌内(IM)给药更容易,并且制药公司在商业上更受青睐。然而,SC和IM施用都受到可以在没有显着疼痛或不适的情况下递送的体积的限制,在前者的情况下,约为1-2ml /注射。尽管如此,已经开发了通过使用水解结缔组织的透明质酸酶来扩增SC空间体积的新技术(Halozyme®Therapeutics)。体积限制与mAbs在水溶液中的溶解度极限(通常为100mg / ml,尽管一些先进的制剂达到150mg / ml)相结合,限制了可通过单次SC施用递送至约 100-150mg的mAb剂量。更高剂量的要求需要增加SC注射的次数。在开发过程中需要考虑的另一个方面是接近100mg / ml的浓度可能导致不可接受的高粘度(> 20cP),这使得制剂难以使用注射器递送。与小分子药物相比,从SC部位到体循环的mAb吸收是一个缓慢的过程,因为它涉及通过淋巴管转移。这种缓慢吸收(t1/2 ~ 2.7天)导致Tmax约为36-72小时。由于淋巴液中的免疫细胞不断对淋巴液进行取样,因此mAb和免疫细胞之间的初始相互作用可导致施用部位处或附近的免疫原性应答。在临床上和临床前在非人灵长类动物中评估的几种mAb的生物利用度为30%至90%。在非人类灵长类动物中观察到的生物利用度通常高于人类;尽管如此,它是一个很好的替代品,通常使用60%的值作为建模的第一近似值。
1.5.2 分配 Distribution
就像吸收一样,mAb在体循环和组织间质中的分布也受其大小的限制。从体循环到组织的mAb的分布是缓慢的过程,通过相对(与小分子相比)显而易见的是,在mAb的双相血浆谱中,末端动力学相缓慢出现。使用放射性标记的111In,Baxter等人证明,在达到假平衡后,约10%的mAb血浆水平分布到组织空间中。观察到该比例在小鼠中相似。此外,由于总间隙体积约为总体水的三分之一(0.6L / kg),因此可以假设每个组织中的间隙空间也约为组织体积的三分之一。因此,预期30%的血浆mAb水平分布到组织间质空间中。因此,如果已知血浆中的mAb浓度,则可以将其用于近似组织间质中的mAb浓度。在诺华公司进行实验证实,在皮肤间质中发现约30%的血浆mAb水平。在该实验中,他们通过灌注真皮间质并测量血浆以及IL17和Secukinumab的间质灌注液来研究Secukinumab,一种用于治疗牛皮癣的抗IL-17单克隆抗体。他们分别在健康和患病个体的真皮间质灌注液中观察到23%和35%的血浆mAb水平。
注意,通过扩散不可能在细胞内分布大小与抗体相当的分子。 通常,细胞表面上的受体与抗体相互作用并将其内化到细胞中。 该过程称为靶向介导的药物处置( target-mediated drug disposition,TMDD),并且负责影响血液中的mAb动力学(详见第4节)。
1.5.3 代谢 Metabolism
具有良好定义(well-defined)的蛋白质结构的生物制剂,不经受基于细胞色素P450的代谢,这对于小分子是常见的。 一般来说,mAb通过三种途径清除:(1)肽酶分解代谢(2)FcRn受体(也称为Brambell受体)再循环(3)TMDD。由于这些生物制剂是蛋白质分子,它们是预期的被蛋白水解酶(如肽酶)降解。
FcRn再循环是一种内源性过程,其调节体内基于IgG的分子的浓度。 具体地,不同细胞上的FcRn受体(例如,血管内皮细胞,巨噬细胞,树突细胞,肝细胞,上皮细胞和肾细胞)在内化时在低pH下结合IgG分子,从而保护它们免于分解代谢。 在内化期间和之后,一部分IgG分子在溶酶体液泡中被分解并被清除,而剩余的部分返回细胞表面并分泌回到间质中。 这延长了分子在体内的停留时间。 许多制药和生物技术公司利用这种机制设计的分子的半衰期比标准IgG分子的通常21天长得多。
第三种清除机制是TMDD。 当mAb与循环免疫细胞,内皮细胞或组织空间中存在的靶标结合时,结合的抗体可以被内化和降解。 mAb的这种耗尽导致血浆水平急剧下降,其特征在于非线性动力学过程的典型凸起形状(详见图5.8和5.4.1.2节)。 在较低浓度的mAb中观察到TMDD,并且随着血浆和间质中mAb浓度的增加,该过程变得饱和,之后抗体动力学变为线性的。 这与小分子形成鲜明对比,小分子在较高的药物浓度下观察到可饱和的动力学行为,其中清除机制已经饱和(通常是基于细胞色素P450的酶)。
1.5.4 排泄 Excretion
通过蛋白水解降解mAb产生的大多数肽和蛋白质,被身体同化并被利用。 在某些情况下(MW <60kDa),这些分解代谢的片段可以通过肾脏排泄。 关于ADME特性的更详细的论文在Shi【20】的综述中提出。
二、使用系统药理学模型进行目标选择和验证
定量系统药理学(Quantitative systems pharmacology, QSP)模型在药物发现的所有阶段越来越多地用于制药行业,因为它们为决策提供了定量和综合的视角,而不是经验观察和定性推理。具体而言,QSP模型利用系统生物学方法来识别和验证治疗干预的新靶点,阐明新药和现有药物的作用机制,并创建指导复杂网络调制的知识库。例如,图5.3描述了控制人体血液凝固过程的凝血途径。已经针对几种疾病开发了这样的模型,例如乳腺癌,心律失常,胰岛素抗性和抑郁症,并且已经在药物发现和开发的各个阶段提供了价值。 ErbB网络模型证明了QSP建模对生物药物发现的影响的一个很好的例子,该模型导致MM-121的发现,MM-121是一种在小鼠肿瘤异种移植模型中显示出强效作用的单克隆抗体。类似的建模方法也导致了MM-141的开发,这是一种靶向IGF-IR和ErbB3的四价双特异性抗体 。 QSP模型也可以通过强大的生物标记策略帮助设计临床试验。最近,为该途径开发了QSP模型,以更好地理解各种网络组分的体外调节对凝血的影响

尽管有其承诺,但QSP建模面临许多挑战:不确定的网络拓扑,有限的定量数据,大部分未知的参数空间以及实验上不可观察的状态变量。 通常,可用的实验数据仅允许估计几个参数,使许多关键参数无法识别。 即使用于数据生成的实验分析,确定最佳采样和给药策略也是一个挑战。 然而,必须通过开发数学模型继续努力对生物过程进行定量的系统级理解。 在下一节中,讨论了一个简单的绑定模型,可用于指导mAb先导物优化的过程。
三、单克隆抗体先导物的优化 MONOCLONAL ANTIBODY LEAD OPTIMIZATION
mAb的成功开发主要取决于其PK半衰期,对靶标的选择性和亲和力,生理化学稳定性,降解机制和免疫原性风险。 由于抗体工程技术的进步,许多这些mAb特性可以定制以满足特定要求。 本节主要关注mAb的PK/PD特性的优化,即PK半衰期和目标亲和力。 下面讨论的数学分析的细节将在别处提交(准备中的手稿)。
在药物发现过程早期,确定mAb的最佳PK/PD特性是至关重要的,因为它为抗体工程组提供了足够的时间来设计,生产和测试mAb。 然而,定义最佳mAb特性的标准需要关于组织定位(tissue localization),周转率(turnover rate)和靶标丰度的系统特异性知识。 一般而言,系统相关信息可以通过实验和文献获得,但在缺乏(可能是新目标)的情况下,可以基于与已知目标的相似性来做出合理的假设。 例如,可以假设相同家族的可溶性细胞因子具有相似的特性。 虽然这种方法不是确定的,但它代表了新的目标的合理起点,可以在有更多信息时更新。
mAb的功效取决于其在体内对靶标的占据,其对靶途径的依赖性以及该分子是否旨在是激动剂(agonist)或拮抗剂(antagonist)。 控制mAb实现所需占据(occupancy)的能力的因素可分类如下:
- 系统相关
- 目标丰度 Target abundance (Rtot)
- 目标周转率 Target turnover rate
- 药物相关
- mAb浓度 mAb concentration (Dtot)
- mAb与其靶标结合的平衡解离常数(KD)
系统相关因素主要由疾病状态驱动,不能修改,但他们的知识允许根据要求优化单克隆抗体的PK半衰期和KD。 通常,临床mAb具有充分表征的PK特性,其反过来决定剂量浓度。 下面,在上面列出的四个因素中的三个之间得出关系:Dtot,Rtot和KD。 为简单起见,忽略了靶标的合成和降解以及mAb和mAb-靶标复合物的消除,因为这些过程倾向于在更慢的时间尺度上发生。 结果,该系统反映了体外的条件。 此外,假定mAb与靶标的结合处于快速平衡状态。因此,
$$ K_{D} = \frac{[D][R]}{[DR]} \qquad \qquad \qquad (5.1)$$
另外,由于质量守恒,
$$ D_{tot} = [D] + [DR] \qquad and \qquad R_{tot} =[R] + [DR] \qquad \qquad \qquad(5.2)$$
将公式5.2中[D]和[R]的表达式代入公式5.1并重新排列,
$$ D_{tot} = \frac{[DR]}{R_{tot} - [DR]}K_{D} + [DR] \qquad \qquad \qquad(5.3)$$
将上述等式除以Rtot, 并将[DR] / Rtot替换为占用率(occupancy, occ),
$$ \frac{D_{tot}}{R_{tot}} = \frac{occ}{1-occ} \frac{K_{D}}{R_{tot}}+ occ \qquad \qquad \qquad(5.4)$$
在其他地方已经报道了表征Dtot和KD之间对于各种Rtot浓度的双相关系的图。在此之后,通过Rtot标准化后绘制上述关系(参见方程5.4和图5.4),因为它允许更准确地表征相对于目标的药物相关参数。很明显,当KD~0.1 Rtot和(2)与目标占用的要求无关时,出现拐点(1)。当KD> 0.1 Rtot(图5.4中的阴影区域)时,达到所需目标占有率所需的总mAb浓度(Dtot)呈指数增长。剂量与Dtot相关;位于该区域的mAb将需要显着更高的剂量,因此可称为次优。具有KD = Rtot的次优mAb将需要~10Rtot的剂量以实现90%的靶标占据。另一方面,对于其目标亲和力已经增强使得KD = 0.1Rtot的mAb,实现类似目标占有率所需的剂量是~Rtot。亲和力的进一步改善,即KD <0.1 Rtot,不会导致成比例的剂量减少,因此mAb可被认为是最佳的。
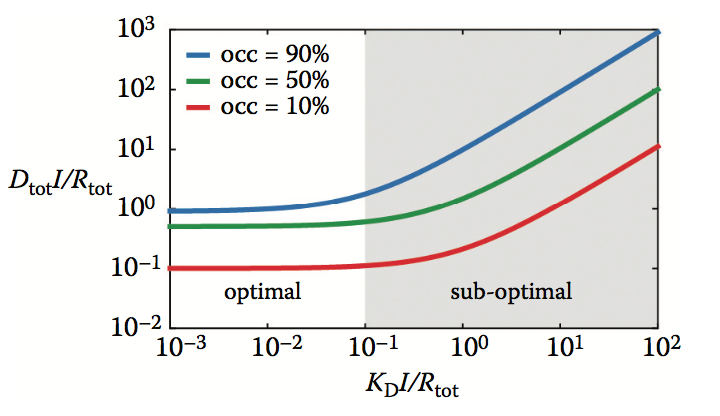
图5.4 Dtot / Rtot和KD / Rtot(公式5.4)之间的双相关系为10%,50%和90%目标占用率(occ)。 阴影和非阴影区域分别代表双相关系的线性和恒定相位。
鉴于对目标占用要求的一些基本了解,抗体工程团队通常面临优化KD或PK半衰期的选择。 对于次优的mAb(图5.4中的阴影区域),达到90%目标占用率(dose90)所需的剂量不受PK半衰期的影响,但通过增加亲和力显着降低(参见从A点到B点的箭头) 在图5.5)。 相比之下,对于最佳mAb(图5.5中的无阴影区域),dose90只能通过增加PK半衰期来减少(尽管略有增加)(参见从B点到C点的箭头)。 尽管PK半衰期的改善可能不会显着降低剂量,但它们确实提供了较少频率给药的优点。 请注意,上述结论与目标占用要求无关。
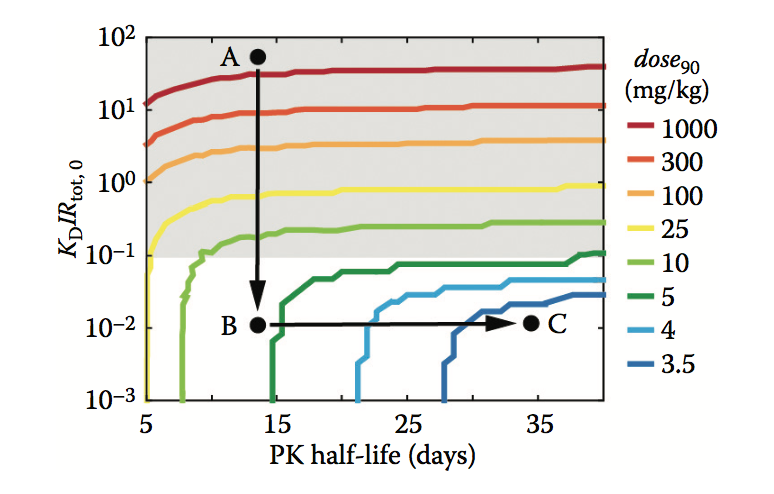
图5.5 优化mAb的PK/PD特性。 等高线图显示了修改mAb特性的影响,即PK半衰期和相对于目标水平的亲和力(KD / Rtot,0),对于达到90%目标占用率的每月剂量(dose90)所需的剂量方案。 阴影和非阴影区域的含义与图5.4中的相同。 模型参数:kelR = 16.4 day-1,kelDR = kelD,kon = 86.4 nM-1-1,koff = kon * KD,ksynR = kelR * Rtot,0。 注意:该模型反映了体内条件,因为它解释了靶标合成和mAb,靶标和mAb-靶标复合物的消除。
接下来,评估KD和dose90之间的关系,以评估目标性质的影响,例如基线浓度( Rtot,0 ),消除速率( kelR)和可溶性与膜结合特征。为清楚起见,可溶性和膜结合靶标的 Rtot,0 和 kelR 分别定义为S0和M0,以及kelS和kelM。使用可溶性靶模型的模拟(图5.6a)显示,降低KD导致较低dose90直至阈值(图5.6b中的虚线),低于该阈值时没有剂量减少。此外,该阈值随着可溶性靶标的基线浓度而增加(图5.6b),这种关系也适用于膜结合靶标(未显示)。有助于阈值定位的另一个因素是mAb-靶复合物的周转。对于膜结合靶标(图5.6c),假定mAb-靶标复合物的周转等于游离靶标的周转,因此,阈值也受靶标消除的半衰期控制(图5.6d)。
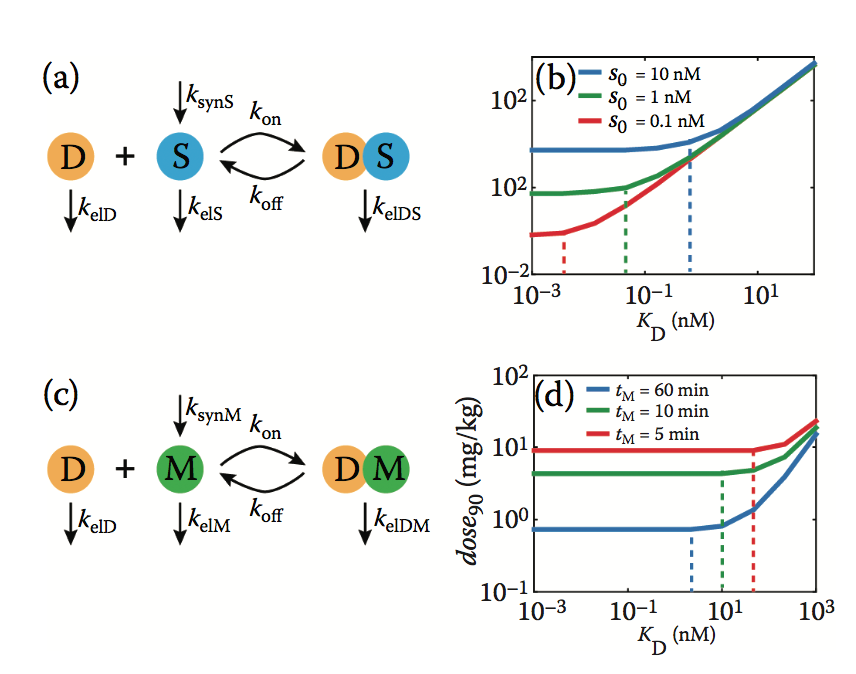
图5.6 mAb的最佳 $K_{D}$ 取决于其与靶标的相互作用。 (a)示意图显示mAb(D)与可溶性靶标(S)的相互作用。 模型中包括的各种过程是mAb与靶标的可逆结合( $k_{on} = 86.4 nM^{-1} day^{-1},k_{off} = k_{on} * K_{D} $),靶标的合成( $k_{synS} = k_{elS} * S_{0} $ ),以及mAb的消除 ( $k_{elD} = 0.03 day^{-1} $),靶标( kelS = 16.4 day^-1 )和mAb-靶标复合物(kelDS = kelD)。 (b)表征剂量达到90%目标占有率(dose90)与不同基线目标浓度(S0)的KD之间的非线性关系的图。 (c)示意图显示mAb(D)与膜结合靶(M)的相互作用。 模型过程与(a)中的相同,具有以下参数的差异:ksynM = kelM * M0,M0 = 1nM,kelM = 0.693 / tM,并且kelDM = kelM。 (d)表征剂量90和KD之间的非线性关系的图,用于膜结合靶的消除的不同半衰期(tM)。
四、人类PK,目标占有率,以及FIH试验开始和有效的剂量
it’s the dose that makes poison — Paracelsus
毒理学的创始人帕拉塞尔苏斯(Paracelsus)在十六世纪提到:dosis facit venenum(“剂量导致毒性”)。这个具有百年历史的报价真正引起了现代药物发现和开发过程的共鸣,在这一过程中,随着主要候选者进入临床测试,安全性和功效之间需要保持良好的平衡。为了确保成功的临床试验,候选药物应在一个剂量范围内进行测试,该剂量范围最大化观察治疗效果的可能性,而没有任何安全问题。因此,合理选择起始剂量和随后的剂量递增方案至最高剂量是新生物实体( new bio- logical entity,NBE)可开发性的关键。在以下部分中,给出了用于预测NBE的人PK的当前方法。这些方法允许对暴露参数的稳健预测,例如最大药物浓度(C max),曲线下面积(AUC)和给定剂量下的最小药物浓度(C min)。随后,详细讨论了用于预测人体药效学(PD),目标占有率和有效浓度的定量模型。最后,基于最小预期生物效应水平(minimum anticipated biological effect level, MABEL )和目标占用率(target occupancy)概念,提出了选择FIH起始剂量和最大剂量的合理方法。
4.1 预测人类PK
4.1.1 线性PK模型
大多数mAb和蛋白质生物治疗剂表现出作为剂量函数的线性PK。在临床前物种(例如大鼠,小鼠或猴子)中获得的PK数据通常通过描述在血浆或血清暴露中观察到的单指数( monoexponential ),双指数(biexponential)或多(multiexponential) profiles的简单数学模型来建模。根据观察到的相数,PK数据可以通过适当的隔室( compartmental )模型进行测量,如图5.7a和b所示。这些模型本质上是经验性的,包含与体内药物分布和清除过程相关的参数,这些参数是根据临床前研究中PK研究的暴露数据估算的。最后,使用异速生长缩放技术( allometric scaling techniques )来缩放这些参数以预测预期剂量下的人体暴露。 Mordenti及其同事[48]是第一个探索清除率和分布容量的研究,并且显示对于每种蛋白质,数据用异速生长方程( $ Y = aW^{b} $)很好地描述,其中a是异常系数 ( allometric coef cient ) ,b是异速指数 ( allometric exponent ),W是体重。基于该数据集,清除参数为0.65 <b <0.84,稳态下的分布体积为0.83 <b <1.05。随后,其他人对15种蛋白质药物,34种治疗性蛋白质,14种单克隆抗体的大数据集进行了类似的分析。最近的研究包括邓等人和Oitate等的研究,利用非房间分析估算间隙和其他参数。虽然这是一种适用于任何PK方法的一般方法,但人们已经充分认识到mAb和许多生物制剂表现出双重反应,表明分布快速期 ( rapid distribution phase ),然后是长末期 ( a long terminal phase )。该领域需要更多的奖学金来探索2区PK参数的缩放。
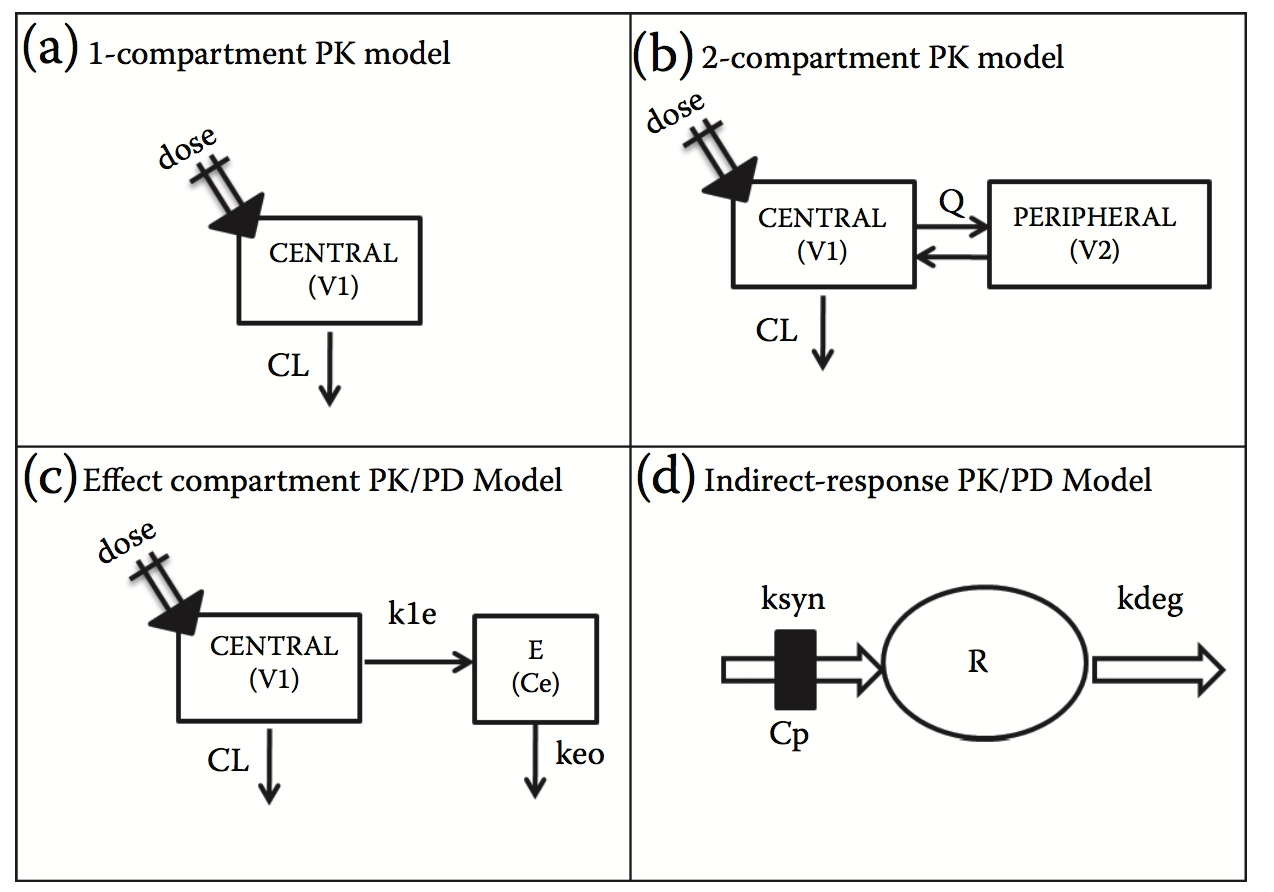
图5.7 PK和PK/PD模型的代表性实例通常应用于药物发现和开发中。 (a)一室PK模型。 (b)两室PK模型。 (c)与描述药物分布延迟相关的PK / PD模型。 (d)间接反应PK / PD模型,其结合药物对受体合成速率的影响。
4.1.2 TMDD模型
虽然大多数mAb和蛋白质生物治疗剂在治疗剂量范围内显示线性PK,但是由于TMDD,目前市场上的许多生物制剂表现出非线性PK。图5.8显示了具有TMDD特征的抗体所见的典型非线性PK谱。如图所示,在较低剂量下观察到更快的清除,因为通过靶向接合以及随后通过运输至溶酶体的内化和药物 - 靶复合物的降解来清除大部分剂量。在较高剂量下,靶标介导的清除率变得饱和,导致终末半衰期延长。 TMDD的大小在很大程度上取决于靶标表达和药物 - 靶标复合物的转换率。对于TMDD的生物制剂,人类PK的投射可能具有挑战性,特别是在起始剂量时,血清暴露不足以完全达到使目标饱和。最近的一份报告通过提出一组转化规则来解决这个问题,这些规则可以应用于猴子数据以预测单克隆抗体的人类PK。这些规则提供了关于如何根据猴子数据建模来扩展药物或目标相关参数的具体指导,使用描述药物 - 靶标结合和各种转换过程的模型。作为该方法的一个例子,图5.9给出了一个TMDD模型(假设准稳态近似),用于基于食蟹猴和恒河猴PK数据的来预测抗TrkB mAb(称为TAM163)的人PK。可用的临床前数据允许估计药物相关(例如,药物消除和分布速率常数)和靶标相关(例如,受体表达)参数。此外,开发了体外测定以估计结合速率常数,kon和koff,以及受体转换参数,kdeg和kint。这样的实验测量允许TMDD模型的完整表征,其反过来可以用于人类投射。充分表征的TMDD模型特别有益,因为它提供了关于体内受体占有率和将导致特定水平的目标饱和度的临床剂量的信息。另外,可以使用完全饱和目标的高端临床剂量(例如,99%)来测试该机制。
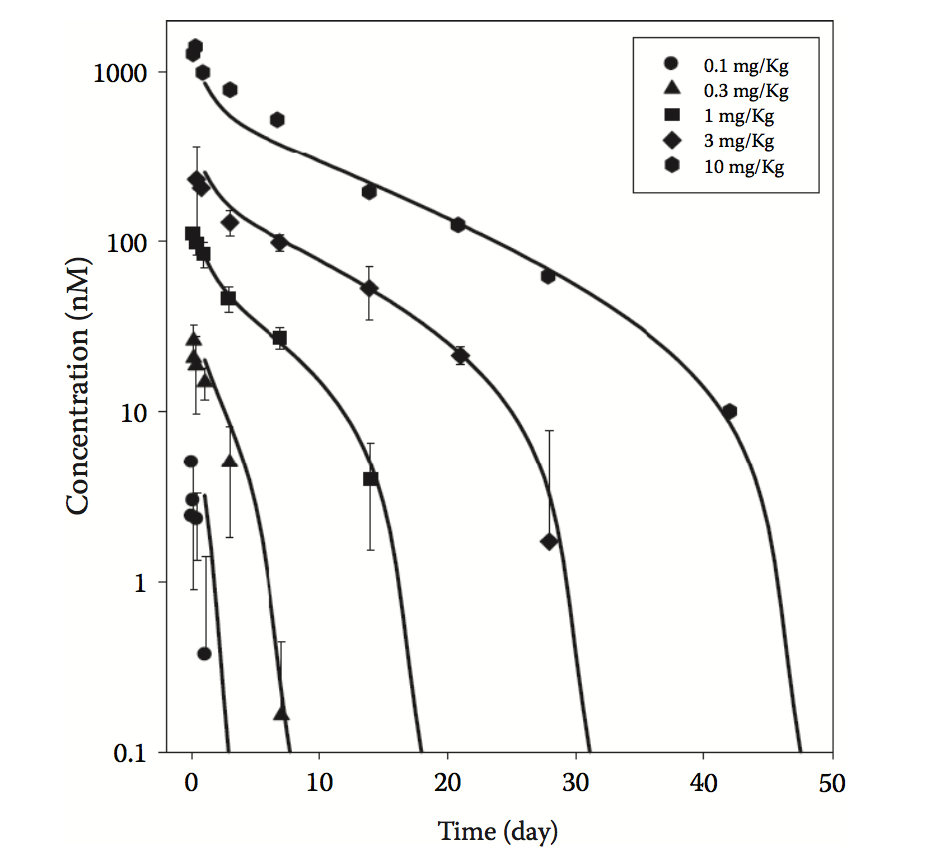
图5.8 抗体的靶介导的药物处置( target-mediated drug disposition, TMDD)行为的特征性非线性PK模型。 符号表示观察到的数据,实线表示使用类似于图5.9所示的TMDD模型的数据的拟合。 剂量静脉内给药。 ( From doi:10.1208/s 12248-014-9690-8. 2015 Mar, 19(2):389–99. Supplemental info. )
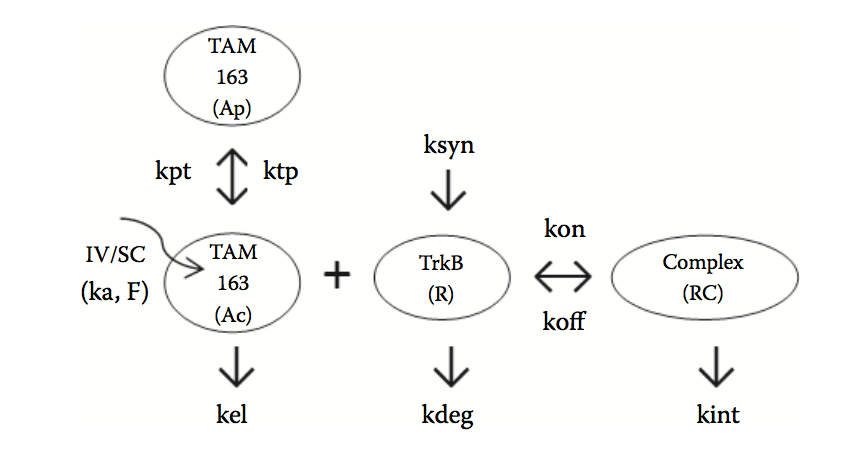
图5.9 描述mAb(TAM-163)与其内源靶标(TrkB受体)结合的TMDD模型结构。在非人灵长类动物中静脉内(IV)或皮下(SC)施用mAb。 kel,kdeg和kint分别代表游离mAb的体内消除速率,游离受体的转换率和mAb-受体复合物的降解速率。 受体的内源合成速率用ksyn表示,而结合动力学由结合(kon)和解离(koff)速率常数控制。 kpt和ktp描述了中心(Ac)和外围(Ap)区室之间的分布过程。 通过SC途径的生物利用度和相应的吸收率分别由F和ka表示。
4.1.3 基于生理学的药代动力学模型
经典的隔室(compartmental)建模将药物分布和消除在各种器官中的复杂过程减少为有限数量的速率常数,这些速率常数通常没有明确的生理意义,除非它们分离成易于解释的量,例如清除和分布容积。此外,尽管简单的区室模型适合于临床前或人类数据并且为临床前或人类数据提供描述性框架,但它们通常缺乏用于将体外数据扩展到体内设置以预测血清或组织暴露方案的机制框架。基于生理学的药代动力学( Physiology-based pharmacokinetic ,PBPK)模型(图5.10a)以定量方式描述人体内的主要生理器官或组织以及它们之间的血液。从它们在环境毒理学研究中的起源到小分子药物分布,PBPK建模方法现在正在应用于生物制剂,最近几个出版物开发了一个平台PBPK模型来描述单克隆抗体和其他生物治疗药物的血清和组织配置。多年来,抗体PBPK模型从经验模拟每个器官到在所有组织中结合IgG-FcRn相互作用并将个体器官分成具有内体交通的血管,间质和细胞亚室,如图5.10b所示

图5.10 描述IgG处置的PBPK模型。 (a)由血浆(实线)和淋巴管(虚线)连接的最重要的器官。 (b)每个器官隔室内的模型结构,包括血管,内体和间质空间(Adapted from Garg, A., and Balthasar, J.P., J Pharmacokinet Pharmacodyn, 34(5): 687–709, 2007.24)
4.2 开始用于FIH试验的剂量预测
选择FIH试验起始剂量的传统方法是基于2005年FDA药物评估和研究中心(Center for Drug Evaluation and Research,CDER)发布的指南,该指南列出了以下关键步骤:
- 在临床前物种中建立无可观察到的不良反应水平(no observable adverse effect level, NOAEL),根据体表面积或体重标准化将其转换为人体等效剂量( human equivalent dose , HED)
- 从最合适的物种(通常是最敏感物种)中选择HED并应用安全因子(至少10倍)以估计最大推荐起始剂量(maximum recommended starting dose, MRSD)
- 基于药理学活性剂量调整MRSD。虽然这些指南很容易遵循,但它们完全基于毒理学研究的NOAEL,主要关注剂量而不是暴露,并且不考虑药物对目标的影响。
在2006年FIH管理TGN1412(CD28超级激动剂mAb)期间出现令人震惊的不良事件后,欧洲药品管理局在新指南中引入了高风险生物制剂MABEL的概念。这些指南强调了一种剂量选择方法,该方法基于对临床前物种的药理学,安全性和疗效数据的综合分析。虽然每个程序都不需要基于MABEL的方法,但是当预期由于夸大的药理学引起的安全问题时,它尤其有用。根据专家科学小组关于第一阶段临床试验的建议,应该使用更广泛的剂量计算方法,该方法考虑了体内动物研究的暴露 - 反应数据以及临床前数据的PK / PD分析。
4.3 对有效或药理活性剂量的预测
过去,基于模拟人类疾病中观察到的病理生物学的某些方面的临床前动物模型,已经预测了人类的有效率(human efficacy)。实例包括胶原诱导的关节炎的小鼠模型,大鼠中的佐剂诱导的关节炎,镰状细胞病的转基因小鼠模型,癌症的小鼠异种移植模型,阿尔茨海默病的转基因小鼠模型和慢性肾病的啮齿动物模型。这些模型的实用性在很大程度上取决于动物与人类疾病驱动因素的相似性以及动物疾病对人类反应的可转移性。而不是仅仅依赖于临床前动物的总体结果反应(例如肾脏疾病模型中的蛋白尿,异种移植模型中的肿瘤大小等),了解通路生物标志物的暴露反应关系对于确定临床前学习(preclinical learnings)如何转化于人类更为重要。暴露和响应之间的关系可以简单到线性或更复杂,涉及反馈环,反弹效应或与蛋白表达相关的转导延迟。图5.7c和d显示了PK / PD模型的两个例子:(1)连接区室模型,其解释了药物进入作用部位(SoA)的延迟,以及(2)间接响应模型,其中预计该药物会影响生物标志物或疾病过程的合成速率。根据观察到的反应,必要的复杂性可以纳入PK / PD模型,以解释数据并提取与药物相关的重要参数(即体内效力或IC50)或系统(即生物标记物合成率,目标周转率等)。这些模型尽管具有经验性质,但在开发用于将临床前数据转化为临床的定量框架以及指导临床给药方案方面可能是有价值的。
由于动物和人类疾病之间的根本差异,动物模型的暴露 - 反应数据可能具有内在的限制。然而,目标占用等客观测量对于预测药物活性剂量非常有用,因为生物制剂与细胞表面或循环中的靶标的结合会触发复杂的下游效应。因此,确定药物管理的目标占用率,在临床前物种中建立占用 - 响应框架(occupancy–response profile),并最终将经验适用于为人类,同时预测人类的药理学活性剂量是非常有用的。诸如SoA模型或最小PBPK模型的新兴想法试图预测作为给药方案的函数的疾病相关组织(即,作用部位)中的目标占有水平。图5.11显示了SoA模型的结构,该模型用于预测脾脏中的目标覆盖率。这些模型解释了生物制剂在组织中的分布,药物与该位点靶标的结合,以及靶标,药物和药物 - 靶标复合物的体内转换。在可溶性靶标的情况下,由于与游离靶标相比复合物的更新周期较慢,观察到总靶标(游离和结合靶标的总和)水平的增加,可以开发生物分析测定法来表征它们的时间过程。随后,可以将这些数据馈送到SoA模型以估计目标占有率,表观结合常数和游离靶标的体内转换率。当有足够的数据时,该模型可以很容易地转化为人类,以模拟各种给药方案和相应的占用率。

图5.11 用于预测组织中目标覆盖的动作站点模型。 kdeg和kdeg’分别是血清(T)和组织(T')中的目标转换率。 ksyn和ksyn’分别是血清和组织中的零级目标合成率。 kel和kint分别是游离抗体和复合物的消除速率。 kpt和ktp分别是从血清到外周和外周到血清的抗体分布率。 kd是平衡结合常数
五、结论和未来方向
虽然制药行业,监管机构和学者越来越多地使用此处描述的PK / PD建模方法,但是一些挑战限制了它们的影响。其中一个主要挑战是人类疾病数据的稀缺性,例如疾病发生和发展的定量信息,生理相关的生物学(例如,组织表达和目标周转率),以及可以预测患者对治疗和住院患者反应的生物标志物,住院患者的变异性。缺乏将动物模型可靠地转化为人类的知识进一步加剧了这个问题。至关重要的是,我们开发成本和时间有效的体外和体内高通量测定和非侵入性成像工具,以便我们可以获得更加定量的人体生物网络图片。随着技术挑战在未来变得易于处理,预计当前PK / PD模型的经验和黑盒特性将让位于更多机制模型,这些模型可以提供目标细胞内途径的多尺度视图,细胞致病性反应,细胞 - 细胞相互作用和器官损伤。这些模型与关于体内药物作用的动态信息相结合,最终将实现药物发现和开发的高效过程。
总结
本章重点介绍了建模和模拟在药物发现和开发各个阶段推进治疗性生物制剂方面的一些关键贡献。具体而言,我们专注于机械PK / PD模型,用于(1)指导选择潜在lead的亲和力要求(2)预测人体药代动力学以设定人体内第一次(FIH)试验的起始剂量,以及(3)项目药理活性或有效剂量,以帮助建立人体机制的证据。本章首先简要介绍单克隆抗体的吸收,分布,代谢和排泄(ADME)特性。随后,我们讨论了可用于研究信号转导途径中抗体依赖性激活或抑制药理学靶标的数学模型。这种相互作用可能导致途径生物标志物的变化,其也可以包括在模型中,从而可以帮助设计有效证明理想治疗结果的临床方案。在适当的地方,我们提供了具体建模方法的详细信息以及选择的示例。
参考资料
- 《Developability of Biotherapeutics》
