【4】生物治疗药物的临床前免疫原性风险评估(Preclinical Immunogenicity Risk Assessement of Biotherapeutics)
一、前言
用作治疗剂的人蛋白质开发的令人惊讶的方面是观察到许多人在患者中引发抗药物抗体( anti-drug antibodies, ADAs)。 临床ADA反应的后果从轻微到严重不等。 抗体可以是瞬时的(transient)或非中和(non-neutralizing)的,在这种情况下,效果可能不是不利的,尽管它们可能影响治疗清除,无论是正面还是负面,导致额外的剂量需求。
当抗体中和(neutralizing)时,这可能会限制有效性,如粒细胞巨噬细胞集落刺激因子(GM-CSF)和IFNβ的情况。在某些情况下,抗体也可识别内源性(endogenous)蛋白质,对患者产生严重后果,如促红细胞生成素(erythropoietin,EPO)与治疗贫血;或血小板生成素(thrombopoietin,TPO)的纯红细胞再生障碍相关,与免疫性血小板减少性紫癜(ITP)和血小板减少症有关。
ADAs也是治疗性抗体技术发展的核心焦点,该技术致力于通过增加候选疗法的人性( humanness of candidate therapeutics ,这个humanness应该怎么翻呢?)来降低患者中ADAs的频率。进入临床使用的第一种抗体之一是完全小鼠抗体muromonab(用于同种异体移植排斥),其在高比例的患者中具有免疫原性(immunogenic)。随后,产生含有与人抗体恒定融合的鼠可变区的嵌合抗体结构域(例如,在iximab和abciximab中),但ADAs仍然以高频率产生。人源化(humanization)技术进一步提高了治疗性抗体的人性(因此鼠抗体的互补决定区[CDR]与选定的框架一起通过从噬菌体展示文库中选择完全人抗体或从人抗体基因转基因小鼠中分离,将残基移植到人种系抗体序列上。虽然这些技术的一般趋势是减少ADA中的ADAs频率。临床上,他们并不常规地提供具有低免疫原性的抗体,例如由完全人抗体阿达木单抗(adalimumab)引起,其中ADAs的发展与缺乏临床反应有关
患者对临床开发中的治疗剂的免疫应答可能导致药物无法进入市场,如TPO和针对A33肿瘤抗原的人源化抗体。因此,免疫原性测试(Immunogenicity testing)被认为是证明临床安全性和有效性的关键组成部分。如果没有免疫原性的临床评估,即ADAs的数据,不太可能获得生物制剂的监管批准,并且FDA提供指导对检测ADAs和风险缓解。如上所述,证据表明,人性( humanness )不是ADA发展的预测因素,并且有许多因素影响ADA是否发展。这些可能与患者,疾病适应症,给药途径或治疗剂的性质和纯度水平有关,所有这些都有助于破坏对人治疗性蛋白质的免疫耐受性。无论促成因素如何,如下所述,引发免疫应答的过程的核心是蛋白质序列内存在人类白细胞抗原(HLA)II类限制性CD4 + T细胞表位,这推动了ADA开发(图4.1),最终形成高亲和力同种型转换抗体,其结合治疗性蛋白质表面上的B细胞表位。因此,用于评估免疫原性风险的计算方法集中在预测治疗性蛋白质中的T细胞和B细胞表位,作为用于铅选择或用于重新设计蛋白质用于表位去除的快速且成本有效的工具。本章重点介绍临床前开发过程中蛋白质的免疫原性的表位预测和评估潜能的不同计算方法。

图4.1 T依赖性抗原的适应性体液反应总结。APC通过非特异性吞噬作用或受体介导的内化作用吞噬治疗性蛋白质。蛋白质通过内体途径中的蛋白酶加工成片段以产生肽片段,其中一些片段可以被MHC II类结合,然后肽-MHC II复合物呈递在细胞表面上。静息的CD4 + T细胞通过T细胞受体结合肽-MHC II复合物,并且在共刺激分子的参与和细胞因子的存在下,被激活。 B细胞还充当APC并且可以在其表面上呈现相同的肽-MHCII复合物,其可以被同源激活的T细胞参与以通过共刺激因子为B细胞提供生长支持。因此,B细胞成熟以产生高亲和力,同种型转换的抗治疗性抗体。该过程的核心是在MHC II类背景下呈递肽并且通过T细胞识别该复合物。 (Modi ed from Baker, M. P., and Carr, F. J., Current Drug Safety, 5(4), 308–13, 2010.23)
二、T-CELL EPITOPE PREDICTION
2.1 T-cell表位的属性
产生高亲和力IgG类抗体的适应性免疫(adaptive immune)应答需要来自CD4 + T辅助细胞的共刺激形式的B细胞的帮助。 T细胞活化的先决条件是在HLA II类背景下呈递来自加工蛋白的短线性肽,可通过T细胞受体(TCR)识别(图4.1)。
存在HLA II类蛋白的同种型:HLA-DM,-DO,-DP,-DQ和-DR。非经典HLA分子HLA-DM和DQ在经典HLA分子-DP,-DQ和-DR.2的肽负载中充当伴侣蛋白。经典HLA II类分子中的表达水平对于HLA-DR是最高的,这可能是它是研究最多的HLA II类同种型的原因。 HLA II类分子由两条链α和β组成,形成两端开放的肽结合沟,允许可变长度的肽结合。这些肽通过氢键以延伸的构象结合在凹槽中。 HLA-DR分子的β链具有高度多态性,迄今为止在全球人口中已鉴定出超过1700个等位基因28,而只有三个α链等位基因。相比之下,HLA-DQ和HLA-DP的α链和β链都是多态的,可能会产生数千种组合;然而,HLA-DQ多样性受到某些α/β链配对的不相容性的限制,并且已发现特定的HLA-DPα/β组合在潜在的谱系中占主导地位。
尽管经典的多形态多样性存在明显的多样性。 HLA分子,肽结合库不是无限制的,并且在HLA类型内和之间的肽结合特异性城市中存在相当大的重叠。肽在结合沟(binding groove)内的结合取决于核心HLA II类结合寄存器内的特定位置(称为锚定残基,termed anchor residues)的氨基酸侧链的性质(包含9个氨基酸)。因此,9-mer结合基序可以从可能与特定HLA II类分子结合的蛋白质序列中确定。然而,肽结合的问题比核心9聚体的分析更复杂,因为HLA II类结合沟是开放式的并且可以容纳不同大小的肽。因此,位于核心9聚体外部的残基(p1,p10和p11)的影响也被认为是重要的。据报道,肽延伸通常导致主要组织的增加。兼容性复合物(MHC)II类分子,具有约18-20个氨基酸的最佳肽长度,35并且在来自树突细胞(DC)的肽洗脱实验中,对于天然存在的肽通常观察到18至25个氨基酸的长度
2.2 计算机模拟(in silico)预测T-cell 表位
2.2.1 HLA Class II Binding Prediction
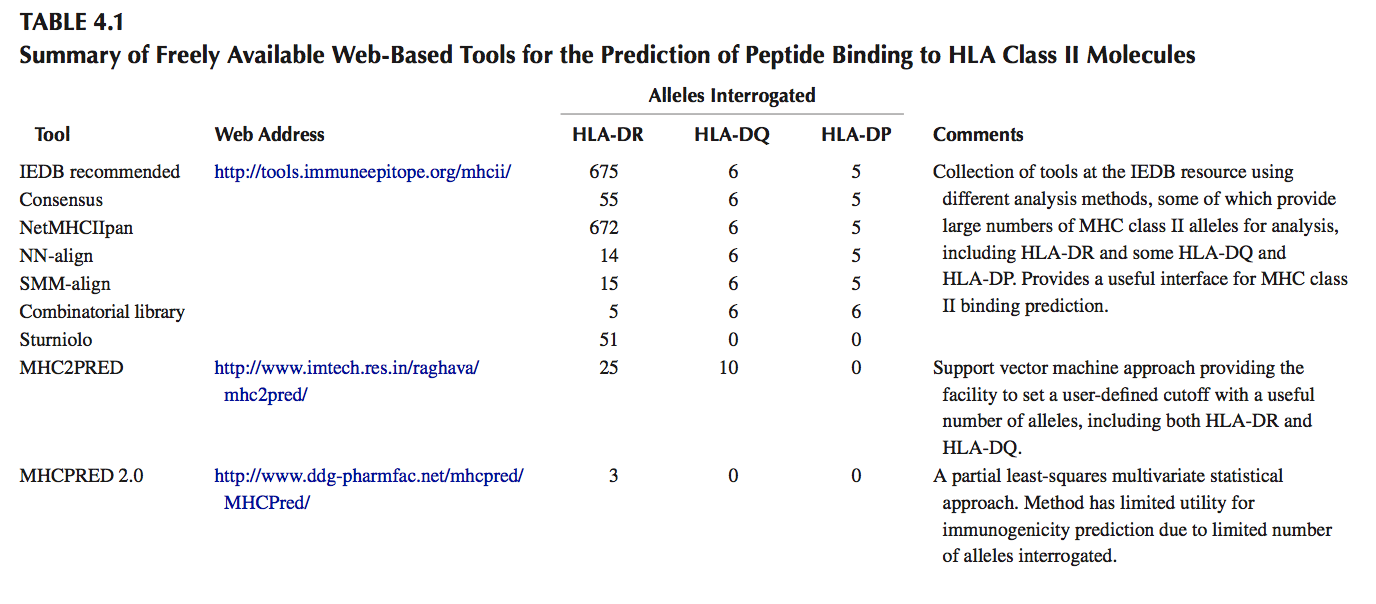
通过体外肽结合实验表征与不同HLA II类分子结合的肽基序,使得能够开发用于预测HLA II类结合肽的计算机算法或结构建模工具(参见表4.1的总结,免费提供基于网络的工具)。 然而,当使用这些工具进行免疫原性预测时,必须考虑许多限制,尤其是所有工具都基于对核心9聚体在HLA II类结合沟内的结合进行预测的事实。 并且不考虑其他交互(如前一节所述)。
计算机算法基于实验肽结合数据的事实导致对算法的预测能力的限制。由于在这些结合测定中仅测试了一小部分天然存在的肽组,因此得到的肽结合数据集是有限的,并且最终使用它作为评分基础的计算机算法也是如此。此外,当比较来自实验性结合测定的HLA II类结合数据时,肽的精确结合寄存器( binding register )通常不清楚,这将影响用于训练计算机算法的数据集的质量。此外,实验性结合分析的特异性和敏感性受试验条件以及试验肽的选择的影响。溶解性差的肽可能不会进入溶液,产生假阴性结果,但由于分子中较大和较可溶的片段的结合以及随后根据“先结合,后修剪模型”修剪较短的肽,可在体内呈现在HLA II类分子上。此外,体内分子伴侣参与编辑抗原呈递细胞(APC)上呈递的肽库,其方式使得具有良好总体亲和力但快速解离率的肽被伴侣HLA-DM释放以促进形成,形成具有长期稳定性的HLA-DR-肽复合物。
计算机模拟HLA II类结合预测算法的另一个限制在于计算肽结合概率的方式。考虑到不同结合口袋的可能性,而不考虑整体结构肽构象。由于计算机工具仅扫描一级氨基酸序列,因此无法评估翻译后修饰或蛋白质质量属性(如聚集)的影响,并且已显示这些因子可在一定程度上改变所呈现肽的数量和类型。最后,这些工具仅预测肽与HLA II类的相互作用,而抗原加工(通过APC)和通过TCR识别肽-HLA II类复合物的关键影响是不考虑。专业抗原呈递细胞内的抗原加工很大程度上决定了潜在的HLA II类肽结合谱的大小,并且在APC细胞表面上呈现该谱后,只有有限数量的肽与T细胞结合。受体具有足够的亲和力或频率来刺激同源T细胞活化。剩余的肽只是HLA II类结合物,由于无反应性(anergy,无反应性)或不存在(通过在中心容忍期间删除)TCR而不刺激T细胞活化 - 细胞室。
总之,这些局限性导致了计算机模拟方法一直过度预测蛋白质序列中存在的实际T细胞表位的数量,因此,最多可以为鉴定具有潜力的绑定到HLA II类的序列区域提供有用的帮助。随后的离体(ex vivo)或体内(in vivo)T细胞活化分析通常需要鉴定真正的T细胞表位,并根据它们诱导T细胞应答的效力对肽进行分级。
尽管存在这些局限性,但计算机HLA II类预测工具对于药物开发过程中的特定应用具有明显的优势,如第2.3节所述。最近对HLA II类肽结合计算评估的最新进展进行了全面评述,这些用于预测免疫原性的工具中最有用的是RANKPEP,PROPRED,Tepitope和NetMHCIIpan,这些算法可以查询广谱人HLA II类等位基因。后者还具有预测与HLA II类等位基因结合的能力,其中还没有任何实验性结合数据可用。已经在免疫表位数据库分析资源(Immune Epitope Database Analysis Resource,IEDB)( www.iedb.org )中选择并策划了一系列工具,为分析蛋白质序列提供了方便的网络界面,并且也提供基于这些工具的共识的方法。同样,iTopeTM软件可查询多达34个HLA II类等位基因,类似于RANKPEP和PROPRED,并将来自体外HLA II类结合研究的数据与通过离体T细胞测定已证明为T细胞表位的大型肽数据库的比对结合起来。
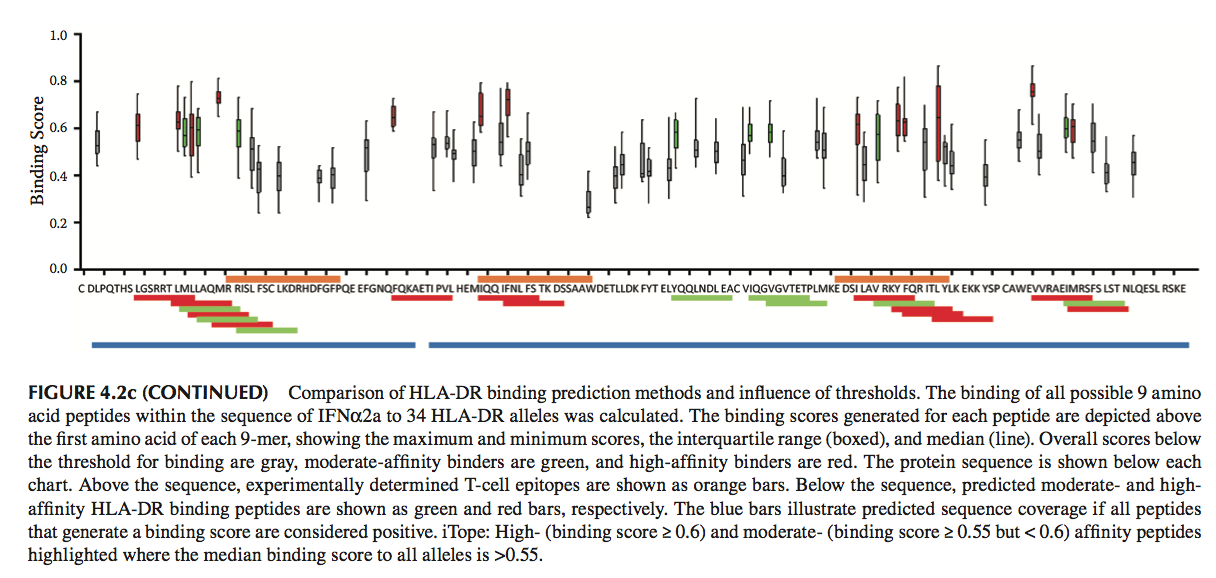
当考虑广泛的HLA II类等位基因时,这些工具的数据输出可能非常复杂,特别是在考虑大量HLA II类等位基因时。为了将数据集减少到可管理的比例并确保预测不跨越延伸的序列片段,根据实验确定的T-cell 表位来设置数据阈值值很重要。这一点由图4.2说明,其中比较了两种算法(NetMHCIIpan和iTope)预测肽与人干扰素α2a(IFNα2a)序列中34个HLA II类等位基因同一组的结合能力。并且将输出进一步与通过离体T细胞测定鉴定的T细胞表位进行比较。图4.2a显示了使用IEDB推荐的阈值的NetMHCIIpan的结果,而在图4.2b中,显示了结合的附加标准–必须生成至少50%的测试等位基因的分数。图4.2c类似地显示了iTope数据,突出显示了高和中结合能力的肽。两种方法之间存在良好的对应关系,其中预测高或中等亲和力结合肽与T细胞表位一致;然而,由于方法的局限性,如上所述,HLA II类结合肽和T细胞表位不总是匹配,并且预测其他结合肽不是实际的T细胞表位。增加NetMHCIIpan的阈值严格性(图4.2b与图4.2a相比)降低了输出的复杂性,同时不影响预测的整体成功。两种方法都说明了设定适当临界值的必要性,其中,如果考虑所有预测的结合肽,则预测几乎整个序列含有重叠的HLA-DR结合肽。

图4.2a HLA-DR结合预测方法的比较和阈值的影响。计算IFNα2a序列内所有可能的9个氨基酸肽与34个HLA-DR等位基因的结合。每个肽产生的结合分数描绘在每个9聚体的第一个氨基酸之上,显示最大和最小分数,四分位数范围(加框)和中位数(线)。低于结合阈值的总分是灰色,中等亲和力粘合剂是绿色,高亲和力粘合剂是红色。蛋白质序列显示在每个图表下方。在序列之上,实验确定的T细胞表位显示为橙色条。在序列下方,预测的中度和高度HLA-DR结合肽分别显示为绿色和红色条。如果产生结合分数的所有肽都被认为是阳性,则蓝条表示预测的序列覆盖。 NetMHCIIpan:中位数分数≤50=高分,中位数分数> 50且≤500=中等分数

2.2.2 Databases of T-Cell Epitopes
科学文献提供了数千个实验鉴定的T细胞和B细胞表位的序列。此外,存在许多手动矫正的数据库,其包括这些先前识别的表位的可搜索记录。这些数据库的一个主要优点是它们包含非常大的数据集,但缺点是包含标准允许包含使用各种实验方法生成的数据,因此具有可变的准确度。到目前为止,这些数据库中最大的是免疫表位数据库(Immune Epitope Database,IEDB)项目。尽管IEDB的大小(目前报告的表位中包含119,185项),但仍然远远不够。实际上,诸如HLA结合和非结合肽数据库( HLA binding and non-binding peptide database, MHCBN)的数据库,其包含HLA结合和非结合肽的矫正数据库,以及已知的T细胞表位,包含IEDB中不存在的序列。 因此,不同的数据库提供了数据集多样性的价值,以及独特的搜索功能,例如识别非HLA结合肽的能力,集成的BLAST搜索设施和HLA疾病关联,所有这些都是使研究人员能够解决特定的研究目标
对于蛋白质治疗,已经开发了已知T细胞表位的特定参考文库,其能够通过BLAST分析筛选新序列,以鉴定在体外T细胞分析中先前已刺激T细胞的同源(或部分同源)肽。
T细胞表位数据库(T-Cell Epitope Database,TCED)包含来自已通过离体T细胞表位匹配分析测试T细胞反应性的肽的数据。 TCED中超过60%的肽来自治疗性抗体可变区序列的T细胞表位匹配(mapping),因此,TCED特别适用于在新抗体可变区序列中搜索T细胞表位的同源性。 。当与传统的计算机HLA II类预测软件结合使用时, BLAST搜索分析HLA II类结合肽、先前鉴定的来自离体T细胞测定的T细胞表位和TCED的序列相似性,可以在分析治疗性抗体序列时降低假阳性的频率。 TCED还包含在体外T细胞测定中测试的肽的信息,其已显示不刺激T细胞活化,并且该信息已被用于构建新的抗体V区序列,例如避免T细胞表位在抗体人源化出现。
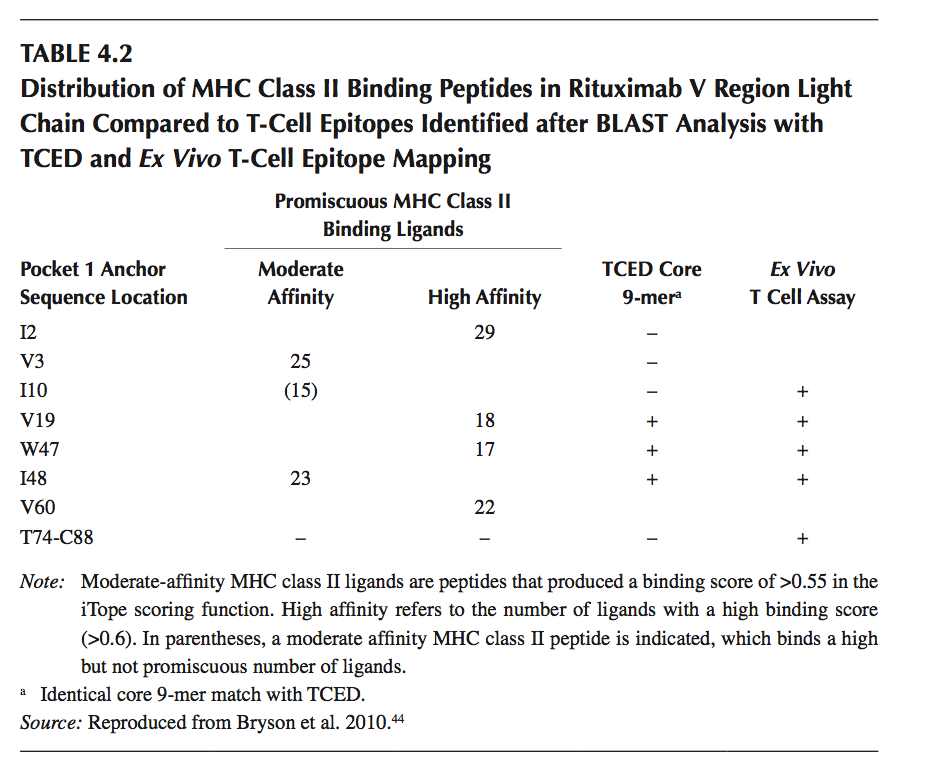
将HLA II类结合预测和V区序列的已知T细胞表位的可搜索数据库组合的能力,与仅通过HLA II类结合预测分析序列相比具有明显的优势。 图4.3显示了鼠嵌合抗CD20抗体(利妥昔单抗,rituximab)的轻链,其使用计算机模拟HLA II类结合软件(iTope)分析混杂的中度和高度HLA II类结合肽的存在。 在利妥昔单抗的轻链中预测了四种混杂的高亲和力HLA II类结合肽和两种中度HLA II类结合肽。

图4.3 鼠嵌合抗CD20 MAb(利妥昔单抗,rituximab)的轻链V区序列的T细胞表位分析。用于预测MHC II类结合肽(iTope)的计算机工具和T细胞表位数据库(TCED)的分析用于通过分析连续重叠的9聚体肽片段来鉴定T细胞表位。混杂的高和中结合力的肽的位置分别用红色和黄色条表示。对于高亲和力结合MHC配体(high MHC ligand,高MHC配体)和中等亲和力MHC配体(moderate MHC ligand,中度MHC配体),显示了34个测试中结合等位基因的数目。 p1锚定残基对于混杂的高中和中等核心9聚体的位置与结合等位基因的数量一致,例如,2位的异亮氨酸结合具有高亲和力的29个MHC II类等位基因,并且是p1锚定残基核心9-mer IVLSQSPAI。与利妥昔单抗序列具有同源性的TCED肽突出显示为绿色条,并且在利妥昔单抗轻链V区序列的离体T细胞表位作图测定中刺激T细胞应答的15-mer肽显示为蓝色条。所提出的TC1和离体T细胞表位作图肽中存在的核心9聚体的p1锚定残基的位置由绿色或蓝色条中的p1表示。 p1表示在体外T细胞测定中重叠的肽分析,其揭示了两种替代p1的位置。 (From Bryson et al., Biodrugs, 24(1), 1–8, 2010)
用轻链V区序列对TCED进行BLAST分析显示与来自不相关抗体序列(其中两个重叠12个氨基酸)的三个15聚体肽序列同源,使用离体T细胞分析,这些序列之前已显示含有T细胞表位(表4.2和图4.3)。四种预测的混杂高亲和力HLA II类配体中的两种与TCED匹配一致。利用重叠的15-mer肽(12个氨基酸)对利妥昔单抗轻链进行离体T细胞表位作图,结果表明跨越TCED鉴定的两个区域的肽刺激了T细胞反应,这些反应是通过增殖和细胞因子分泌来测量的。跨越第三区域的肽也引起离体T细胞应答,其预测不含有大量HLA-DR II类结合配体(尽管该区域可含有HLA-DQ或-DP限制性结合肽)或被鉴定为TCED中的一场比赛。因此,从利妥昔单抗轻链V区序列中,被鉴定为含有混杂的高和中等HLA II类结合肽的四个区域中的两个实际上含有T细胞表位,并且三个表位中的两个被鉴定为离体T细胞表位作图也由TCED鉴定。这些数据显示虽然TCED可用于区分HLA II类配体和T细胞表位,但数据库显然不是详尽的,并且新的T细胞表位可能存在于TCED中未表示的序列中。
然而,由于TCED肽并不总是与测试序列具有完全相同的序列同源性,因此关键HLA锚和TCR接触位置的变化将影响肽刺激T细胞应答的能力。 因此,TCED鉴定的表位可能不总是反映在部分同源肽在体外T细胞测定中刺激T细胞应答的能力中。 为了使这种影响最小化,比较了氨基酸侧链的理化性质,其中核心9聚体的残基在测试序列和TCED中的匹配之间不同。 只有在核心9聚体中显示> 50%同源性并且在非同源锚定位置具有保守残基的核心9聚体被评分为命中。
使用计算模拟HLA II类结合工具和T细胞表位数据库的组合分析T细胞表位的抗体V区序列,缩小仅通过计算模拟HLA II类结合分析确定的潜在T细胞表位的数量,提高了计算机免疫原性预测方法的准确性。然而,所讨论的计算模拟工具的局限性突出了进行额外T细胞表位分析的重要性,例如为了确保在进行临床研究之前选择最佳导联,而进行的药物开发途径中更下游的全蛋白离体T细胞测定。
2.3 用以药物开发的HLA结合预测算法
鉴于预测肽与HLA II类结合的算法的问题(如上所述),需要仔细考虑这些计算模拟工具的应用。计算模拟工具在预测治疗性蛋白质中T细胞表位的精确位置和效力的能力方面的局限性排除了它们在评估先导蛋白(lead proteins)的潜在免疫原性中的用途。然而,已经发现这些工具可用于筛选通常在先发现期间产生的大量序列,其中通过更准确的离体方法进行T细胞表位分析是不切实际的。在这些新序列中HLA II类结合肽的频率和杂乱性可用作标准,以便选择可能含有较少潜在T细胞表位的序列。
由于HLA II类基因座在远交群体(outbred population)中具有高度多态性,导致不同的个体表达不同的等位基因以形成单倍型(haplotype),因此这些工具可用于评估肽与大量HLA II类等位基因的结合,以便可用于评估或去除蛋白质治疗剂的免疫原性,特别是因为不同的HLA II类分子可能具有显着不同的肽结合特异性。一旦候选铅的数量减少,例如,在早期临床前开发期间,可以通过使用MHC相关肽蛋白质组学(MHC associated peptide proteomics,MAPPs)和离体T来确定T细胞表位数量及其效力的更准确评估。
另一个应用领域是通过体外或离体方法分析已经被鉴定为T细胞表位的肽,以鉴定可能对肽-HLA结合重要的关键残基,并选择可能减少或消除相互作用(去免疫)的这些残基的替代残基。 此外,由于已经证明治疗性蛋白质中的单个氨基酸变化具有调节临床免疫原性风险的潜力,因此可以使用计算模拟点突变的人类治疗性蛋白质(引入以改善其性质),作为最初的高通量筛选,以选择具有最小免疫原性潜力的替代品。
三、B-CELL EPITOPE PREDICTION
3.1 B细胞表位的属性
B细胞表位的预测由于大多数表位是构象(conformational)和不连续的事实而变得复杂; 也就是说,它们由蛋白质残基组成,蛋白质残基可能在蛋白质序列中(或甚至在不同的蛋白质亚单位上)远距离分离,但是被二级,三级和四级结构紧密接近。少数B细胞表位是线性的,并且这些表位的预测可用于设计用于免疫策略的肽以产生针对特定蛋白质的抗体。65 已经全面综述了B细胞表位的结构和抗体结合
3.2 计算模拟预测B细胞表位
早期方法依赖于鉴定线性蛋白质序列中具有高亲水性(hydrophilicity),可及性(accessibility)和可移动性(mobility)的区域以获得氨基酸倾向量表(propensity scales),其可用于鉴定极有可能在蛋白质表面上溶剂暴露的肽,因此可被抗体发现。对这些方法的综合分析表明它们的预测值几乎不比随机值好。与预测结合HLA II类的肽一样,使用评估B细胞表位的许多特征的复杂计算技术提高了预测方法的可靠性。例如,Chen等人在将支持向量机(SVM)分类器与各种氨基酸倾向量表组合时声称预测准确度为72.5%。改进的SVM方法能够以76%的准确度预测线性B细胞表位,并且数学形态学方法被证明是有效的,特别是用于预测具有低至中度抗原性的表位。更近期的方法包括多元线性回归工具和基于已知B细胞表位和已知非表位的大数据集的方法。与所有基于序列的预测工具一样,训练数据集的质量和大小对于成功至关重要。
构象表位(conformational epitopes)的预测更成问题,特别是如果抗原的结构未知。 使用结构数据预测任何蛋白质的构象表位的方法包括CEP,DiscoTope 2.0,ElliPro和CE-KEG。还开发了将实验数据与计算结合使用以预测构象表位的方法。 例如,Pep-3D-Search使用模拟表位(模拟构象表位的线性肽)的序列信息,这些信息是通过肽噬菌体展示文库实验生成的,并试图将模拟表位映射到抗原表面。 最近已经回顾了构象表位预测的当前进展,并且提供了可用的基于网络的工具的概述。 最近的发展包括一种预测B细胞表位的准确度大于70%的方法和一种综合多种预测策略的组合方法【78】。
与T细胞表位预测工具一样,已在IEDB(www.iedb.org)中选择并策划了一组B细胞表位预测工具,该工具使用Discotope 2.0和ElliPro提供用于分析蛋白质序列的网络界面。
3.3 用以药物开发的B细胞表位预测的算法
B细胞表位预测的效用主要在于疫苗设计领域,其中可能需要引入或增强B细胞表位; 然而,假设成功提高针对抗原的抗体反应将导致疫苗接种成功,这是免疫反应的过度繁殖(oversimplication)。 预测结合特定B细胞表位的后果,例如病毒中和,在可预见的未来可能是无法实现的。
从工程蛋白质治疗的角度来降低免疫原性的风险,B细胞表位预测已被用于镶面veneering),即通过突变去除鉴定的B细胞表位以试图使蛋白质对抗体不可见并减少 ,消除患者血清中已存在的抗体的交叉反应。 不幸的是,抗体反应是高度适应性的,这种类型的工程可能会导致蛋白质治疗的其他表面区域通过B细胞表位扩散被识别,这一过程已被充分描述为自身免疫性疾病。(这样就尴尬了。。)
四、总结
现在有各种各样的复杂网络工具可用于预测蛋白质序列内的HLA II类结合肽(潜在的T细胞表位)和B细胞表位。 现在可以以中等的准确度进行HLA II类结合预测; 然而,特别是在鉴定T细胞表位方面,所有方法都是过度预测的,因为它们无法模拟参与T细胞表位形成的所有生物学过程,但也可能无法预测与弱结合HLA II类的肽相关的T细胞表位。这种限制影响了这些工具用于分析潜在蛋白质治疗的临床免疫原性潜力的效用; 然而,它们可用于高通量筛选大量候选分子,并用于估计特异性突变对HLA II类结合蛋白的影响。
B细胞表位预测仍然具有挑战性,主要是因为大多数重要表位是构象的(conformational),因此需要了解三级和四级结构。 此外,虽然T细胞表位预测可能允许蛋白质再造去除强有力的ADA反应的发展途径中的关键步骤,但去除B细胞表位可允许工程化蛋白质通过预先存在的抗体逃脱结合,但可能不 防止适应性免疫反应产生新的抗体特异性,其将识别蛋白质的工程化区域或其他表面区域。
讨论
- 为什么分析更多的是分析TCE
- TCE的多个肽段需要合并么?还是计算其亲和力之和就好
参考资料
- 《Developability of Biotherapeutics》
