【5.2.2】获取exons、cds的序列(Genome Browser)
一、网页版的方法
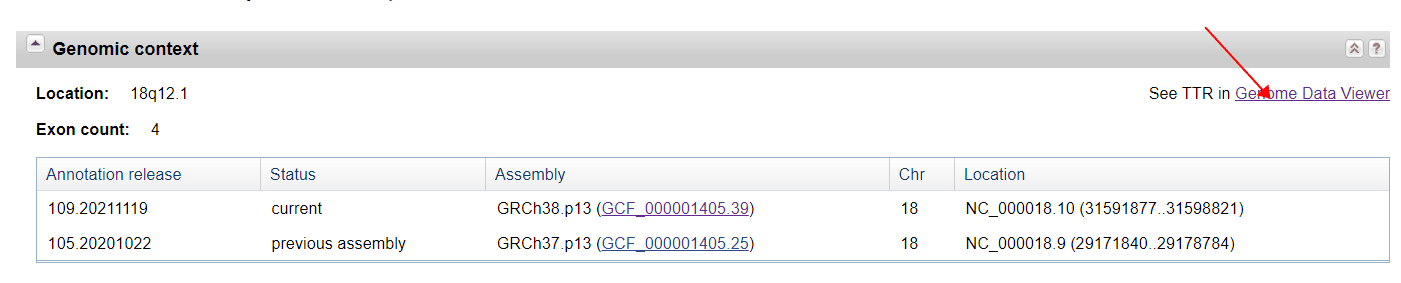
1.先在NCBI上查找对应的基因信息,然后打开 Genome Data Viewer
https://www.ncbi.nlm.nih.gov/gene/7276
2.在Genome browser中分析 ( https://www.ncbi.nlm.nih.gov/tools/sviewer/legends/ 说明文档)
https://www.ncbi.nlm.nih.gov/genome/gdv/browser/gene/?id=7276
可以看到该基因在基因组中的位置。该基因中外显子的个数,可以选择不同的外显子。蓝色的方框为外显子。
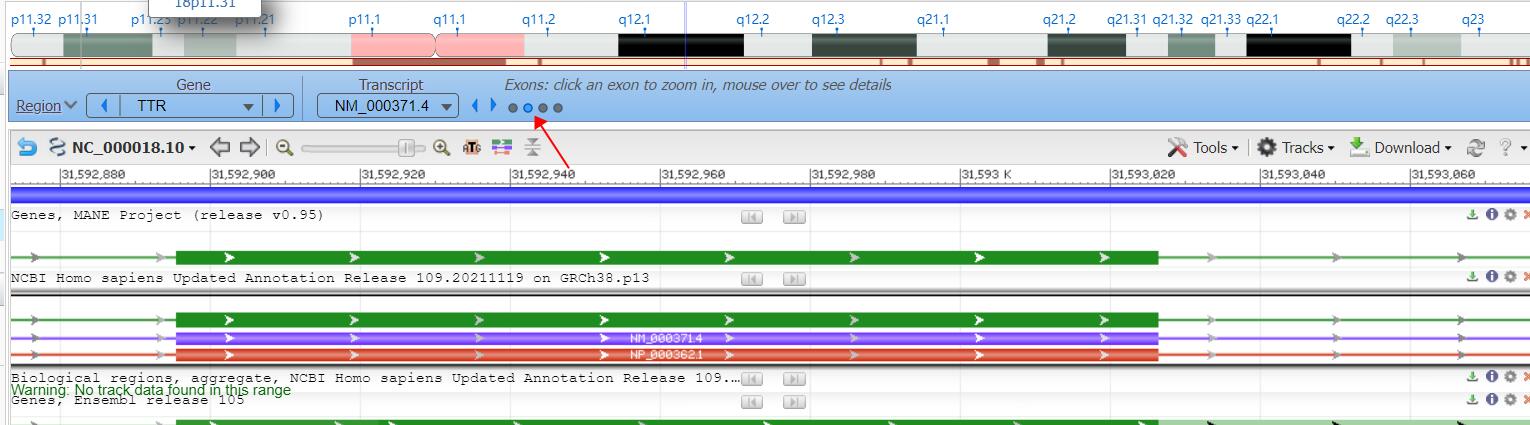
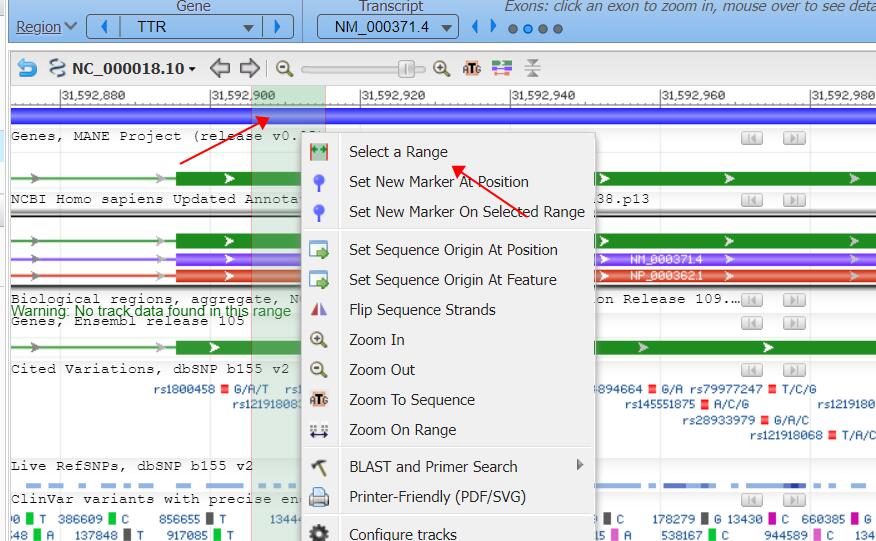
单击右键,选择区域,然后左右拖动,确定选中的位置
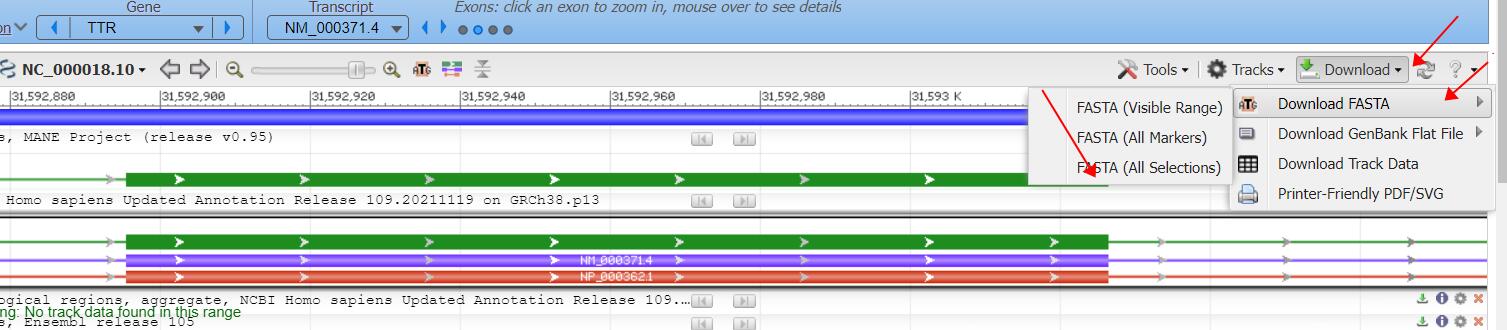
然后,选择下载序列
三、获取人的CDS和exons
使用外显子和内含子分布图重新审视人类基因组。 人类基因组中的 26,564 个注释基因(构建于 2003 年 10 月)包含 233,785 个外显子和 207,344 个内含子。 每个基因平均有 8.8 个外显子和 7.8 个内含子。 每条染色体上大约 80% 的外显子长度小于 200 bp。 < 0.01% 的内含子长度小于 20 bp,< 10% 的内含子长度超过 11,000 bp。 这些结果表明对剪接机制的限制以剪接出非常长或非常短的内含子并提供对最佳内含子长度选择的洞察。 有趣的是,每条染色体上内含子和基因间 DNA 的总长度与确定的染色体大小显着相关,相关系数分别为 r = 0.95 和 r = 0.97。 这些结果表明它们在基因组设计中的意义。
3.1 目前采取的办法
直接根据NCBI的cds序列来提取。
鸡(Gallus gallus (chicken))的CDS 基因序列,但需要根据GENEID来去重
https://www.ncbi.nlm.nih.gov/genome/?term=Gallus+gallus+domesticus
猪(Sus scrofa (pig))的CDS 基因序列,但需要根据GENEID来去重
https://www.ncbi.nlm.nih.gov/genome/?term=pig
wget -c https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/003/025/GCF_000003025.6_Sscrofa11.1/GCF_000003025.6_Sscrofa11.1_cds_from_genomic.fna.gz # 用这个cds序列,根据GeneID保留一个序列
3.1 提取任的外显子基因序列
wget -c https://hgdownload.cse.ucsc.edu/goldenpath/hg19/bigZips/hg19.fa.gz
wget http://hgdownload.cse.ucsc.edu/goldenpath/hg19/database/refGene.txt.gz
只选取外显子:
zcat refGene.txt.gz|sort -u -k13,13|cut -f3,10,11|awk 'BEGIN{OFS="\t"}{split($2,start,",");split($3,end,","); for(i=1;i<length(start);++i){print $1,start[i],end[i]}}' > exons.bed
sort exons.bed |uniq > exons_2.bed
/data/software/bedtools2/bin/bedtools getfasta -fi hg19.fa -bed exons_2.bed -fo exon.fa
只选取CDS
cat refGene.txt |awk 'BEGIN{OFS="\t"}{{print($3,$5,$6,$2)}}' | sort |uniq > cds.bed
/data/software/bedtools2/bin/bedtools sort -i cds.bed > cds.sorted.bed
/data/software/bedtools2/bin/bedtools merge -i cds.sorted.bed > cds.sorted.merged.bed
/data/software/bedtools2/bin/bedtools getfasta -fi hg19.fa -bed cds.sorted.merged.bed -fo cds.fa
2万个cds,即2万个基因
(base) [sam@localhost homo]$ wc cds.sorted.merged.bed
22570 67710 544366 cds.sorted.merged.bed
3.1 提取cds序列
下载 cds的核算序列
http://ftp.ensembl.org/pub/release-104/fasta/homo_sapiens/cds/Homo_sapiens.GRCh38.cds.all.fa.gz
所以最终对Homo_sapiens.GRCh38.cds.all.fa进行处理,同一个起点,保留重点大的那一条cds(用的Python脚本,得到Homo_sapiens.GRCh38.cds.unique.fa)
(base) [sam@c01 homo]$ grep '>' Homo_sapiens.GRCh38.cds.unique.fa |wc
91934 1241603 23001467
参考资料
