6.1 微生物群落和人类微生物组的菌株级流行病学
早在科学界就已经确定了特定微生物菌株的生物学重要性和不同的代谢能力。过去,已经基于微生物分离物对菌株进行了很大的定义和表征。但是,新技术的出现使人们能够评估它们在微生物群落和人类微生物组中的生态和表型。尽管现在更加明显的是,致病性菌株变异如何危害人类健康,但微生物组中微妙的遗传变异的后果直到最近才暴露出来。在这里:
- 我们回顾了菌株的操作定义(operational definitions of strains)(例如遗传和结构变体,genetic and structural variants),因为现在可以使用不同的高通量(通常与培养无关的技术)从微生物群落中鉴定出菌株。
- 我们总结了菌株在人体中的分布和多样性,以及它们与健康维护,疾病风险和进展以及对摄食(例如饮食或药物)的生化反应的新兴联系。
- 我们列出了利用高通量测序以及其他分子和“培养组学”技术鉴定,定量和追踪菌株的方法。
- 最后,我们讨论了群落研究在弥合实验差距和更好地了解菌株对人类微生物组健康影响方面的意义。
一、前言
早在十九世纪,就已经认识到特定微生物菌株的表型和生理特征的重要性。例如,罗伯特·科赫(Robert Koch)的假设是区分引起疾病的“病原体”(pathogens)和良性但密切相关的微生物变体[1]。因此,几个世纪以来,在其他方面相似的微生物菌株之间出乎意料的差异在传染病管理和微生物学中至关重要,但直到最近才在微生物群落及其生态学方面变得可访问。在群落内检测和区分密切相关的微生物菌株仍在技术上具有挑战性,我们将在此讨论几种高通量培养无关和基于培养的方法。不过,更重要的是,这项工作的开始表明,人类微生物组中的菌株变异对我们“正常”微生物居民的结构,功能,免疫学和流行病学的影响与致病性的定义一样重要(专栏1 )。
Box1:
令人惊讶的是,没有关于微生物菌株(microbial strain)(或就此而言的物种,species)构成的通用定义[2,3]。造成这一困难的因素很多,包括:
- 微生物进化的速度
- 许多微生物基因组的可塑性
- 流动元素的流行和侧向转移( the prevalence of mobile elements and lateral transfers)
- 很难通过非分子方法区分许多微生物类群或进化枝,
- 以及微生物学和微生物系统学的整体自然史。
这种歧义导致了一个领域,即同一物种的不同微生物菌株可以相差多达5%的核苷酸同一性,或者其基因含量的30%或更多[4]。因此,即使在表观上相似的良性,微生物相似的微生物菌株变体,与大多数真核生物种相比,在基因组上的差异也更大,并且大多数相关术语可以取决于上下文或在操作上(operationally)定义:
物种(Species):微生物物种的定义基于
- 全基因组或全基因组核苷酸或氨基酸系统发育同一性阈值;
- 总体微生物生理学/形态学/表型;
- 微生物在其宿主或环境中诱导的表型(例如,人类病原体);
- 微生物的宿主或环境,例如特定的地理或生化来源[5]。
因此,必须不断地针对新的,新出现的分子和表型信息解析微生物系统的100多年历史,从而导致对微生物物种的操作定义大致分为两类:
- “在先前某个时间点被定义为物种的进化枝”(clades defined as species at some previous point)
- “符合特定定量系统发育标准的包层” [6]。(clades that meet specific quantitative phylogenetic criteria)
如果系统发育(基因型)被认为是一种特征(即表型),通过它可以将分离株或群落成员分类为自相似组,那么这两个定义可以被认为是大致相同的。
物种组或复杂物种(Species group or complex):根据基因组或其他标准[7],没有经过很好的区分的一组分类学定义的物种。这些通常是由于后来被证明是(基本上)同一生物的多个独立鉴定而在微生物系统中出现的。相反,如果在分类学上确定的单个微生物物种,例如,最初没有通过生理学进行区分,但后来发现在分子上是不同的,则可以证明它们代表隐含的复合物( implicit complexes )。
亚种进化枝(Subspecies clade):在群落中,在operationally上界定为一组的相关生物或系统发育差异半径小于并包含在一个亲本物种之内[8]。这使得群落内的微生物基因型可以独立于其潜在系统进行操纵,因为例如,某些分类学上定义的物种可能无意中捕获了广泛的基因型(因此可以使用多个亚种进化枝更好地描述它们),而另一些则可能被证明是密切相关或近乎相同的(因此更好地描述为单一物种复合体)。从历史上看,亚种也曾指某个物种内表型上不同的基团[5],它们可能是单系统的,也可能不是单系统的。
分离株(Isolate):经过推定和/或菌落采摘等过程后,通常在体外(例如在单培养中)进行无性(例如大概)生长,测定和操作的推测克隆菌株[9]。根据Bergey手册[10]等规范参考,如果没有基因组定义,分离株通常根据表型(如形态)进行区分,例如:中等特异性(medium specificity);血清学,噬菌体或细菌素敏感性;生化反应;致病性或其他微生物生理学。
菌株(Strain):从历史上讲,这意味着要分离微生物,尽管该定义不适合微生物群落研究。在这种情况下,该术语被不同地用于指代特定的微生物基因组或克隆相同细胞的集合(即基因型)。来自同一祖细胞的一个或多个菌落(被认为是);或实际上,通常是在相对较小的系统发育变异范围内(即非常窄的亚种进化枝)内的一组细胞或基因组。
特别是在定义上是异质细胞(heterogeneous cells)集合的群落内,事实证明,在包含此类密切相关但高度可变的基因组的细胞之间进行检测和区分在技术上具有挑战性。确实,目前尚不清楚大多数微生物谱系在典型的体内群落中如何克隆。这既提出了有关在任何微生物群落中密切相关的基因组变异的产生和维持的基本问题,也提出了有关人类微生物组中菌株的个性化和健康后果的翻译问题。由于即使是密切相关的微生物菌株之间在遗传和基因组(即功能)上的广泛差异,迄今为止,很少有人将“普通”微生物菌株与其健康后果相关联的工作[11,12,13,14]。因此,在这里,我们回顾了迄今已知的人类微生物组中携带的微生物菌株变异体的生态学和影响,检测和流行病学的定量方法,以及潜在的后续步骤,包括表征其惊人的大的生物化学暗物质的泛基因组含量。
二、人类微生物组的群里规模调查显示,健康与疾病中意想不到的微生物菌株多样性
数十年来,基于培养的分离物比较遗传学一直是微生物表征的主要手段,并且与培养无关的技术一起,在高通量“培养学”和创新性分离方法的时代中,它变得越来越重要[15,16]。特别是对于既具有临床意义又相对易于培养的人类病原体,在某些情况下已使用数百或数千个基因组来比较菌株及其传播,将SNV和结构变异与微生物或宿主表型相关联,并确定遗传和进化物种和其他进化枝的architectures [17,18,19]。Metagenomic 学方法具有独特的能力,可以将这些针对菌株的研究扩展到几乎任何环境或微生物,同时利用分离基因组学已经积累的见解。特别是,如果“菌株”( strain )被认为是克隆基因型,则它必须对应于一组特定的基因和所产生的功能。这种对菌株的功能性观点已涵盖了广泛的操作架构( operational architectures ),因为某些过程在整个进化枝中都得到了很好的保守(例如,在Faecalibacterium prausnitzii中生产丁酸盐[20,21])。相反,其他细菌甚至在特定的良性或致病性物种中也是高度可变的,肠道中的大肠杆菌是最突出的例子
2.1 人肠道微生物组中的菌株
肠道是人类微生物组中最大的生物量储存库,是人体最大的免疫暴露场所,也是微生物组相关疾病研究最深入的贡献者,也是与人类相关的生态多样性最丰富的微生物栖息地之一[23]。 这也是密切相关菌株之间微生物生理学根本不同的几个最典型例子的来源,例如与肠道出血性大肠杆菌O157:H7等急性病原体相比,大多数肠道中携带的良性大肠杆菌变异体,长期风险,例如大肠癌与pks + +大肠杆菌中大肠杆菌素的产生有关[25]或益生菌大肠杆菌Nissle 1917 [26]。 分离培养物已鉴定出与进化优势相关的其他菌株特异性特征,包括增加的毒力[27],流动性[28],营养获取,抗生素抗性[29]和防御[30]。
婴儿肠道中丰富的菌株很少在母体微生物群中丰富[31,32,33,34],并且通常在生命的前1-2年内被替换[35,36]。它们与母体,家族或一般环境菌株的相似性本身也是高度可变的且具有物种特异性[31、32、37],但即使是微小的结构变异也可能在时间特异性发育窗口期间对免疫程序至关重要[38,39,40] ,41]。像人类基因产物的发育变体(例如血红蛋白形式)[42]一样,这种生命早期活力具有功能性后果:例如,长双歧杆菌被选择用于母乳喂养婴儿的人乳低聚糖(HMO)[43],而紧密结合成年肠道中相关的长双歧杆菌菌株通常具有发酵碳水化合物的能力,但不具有HMO的发酵能力[44]。婴儿肠道中丰富的菌株在母体微生物群中很少出现[31,32,33,34],并且通常在生命的前1-2年内被替换[35,45],但即使是很小的结构变异也可能在生命中至关重要。在特定时间的发育窗口期间进行免疫编程[38,39,40,41]。最终,微生物菌株变异不仅影响宿主和个体微生物的生理,而且影响整个肠道菌群的生态和系统发育:幽门螺杆菌是与人类宿主群体相似的居民微生物遗传变异的最著名例子之一[46]。 ],但最近已证明肠道微生物组的多个子集就是这种情况,例如普雷沃氏菌[12]或直肠真细菌[47]。这导致肠道微生物群落菌株的进化和多样化与宿主迁移,地理和生活方式之间的联系[8,48]。
在肠道中与此相关的最关键的环境因素之一是饮食,无论是急性饮食还是超过进化时间尺度。然而,由于测量多种人类饮食,长期饮食与其他环境因素的混杂以及饮食与微生物生化相互作用的复杂性等挑战,在人类人群中很难理解这种关系的细节。的确,饮食仅代表肠道微生物与我们的生化环境相互作用的一个方面,迄今已发现了一些例子,这些例子包括地高辛[49],二甲双胍[50],对乙酰氨基酚[51]等潜在的菌株特异性代谢。 关于饮食本身,De Filippis等人,例如,[53]发现参与者中更多的体育粉虱更紧密地遵循地中海式饮食,其中富含橄榄油,鱼类,水果和蔬菜。相反,Kovatcheva-Datchary等 [54]观察到,即使在相同的大麦富含饮食下,Prevotella也仅在特定参与者中富集,可能是以菌株特异性的方式。 De Filippis等。 [55]后来发现低脂饮食个体之间的异质性相似。其他例子包括产生短链脂肪酸(SCFA)的细菌菌株,这些细菌对富含纤维的饮食有不同的反应[56,57]。肠道中与饮食相关的菌株特异性的最极端例子之一是益生菌,如乳酸杆菌和双歧杆菌,其中发酵食品具有的特征性菌株与人类肠道中更常见的菌株高度不同[58]。益生菌的健康后果也可能因菌株而异,取决于输入的微生物组的菌株情况[59],也取决于益生菌的菌株,例如,最近提出的一些双歧杆菌促进癌症免疫治疗的能力[60]。 ]。
2.2 肠道菌群菌株是胃肠道和全身性疾病的危险因素
虽然许多研究已将总体微生物组结构或微生物种类的丰富与胃肠道(GI,gastrointestinal)或系统性疾病联系在一起,但很少有人发现与这些疾病相关的菌株特异性微生物变体。就微生物组而言,炎症性肠病(IBD)是研究最深入的慢性胃肠道疾病之一,在IBD中,大肠杆菌和鲁米诺球菌的亚种均与疾病的严重程度有关[61,62]。霍尔等。 [13]注意到在IBD肠道中,R。gnavus菌株的一个特定亚群更为丰富,其丰富性可以适应氧化应激反应,粘附以及铁和粘液的利用。脆弱拟杆菌菌株在IBD小鼠模型中表现出不同的行为,导致差异性IgA诱导[63],并已与宿主在单菌落化中的免疫调节作用相关[64]。尽管有数十年的工作证明了这些变体在动物单克隆化过程中的作用,但要了解它们在人肠道中的作用仍然具有挑战性,因为对于大多数微生物群落遗传变体而言,这相当于一项全基因组关联研究(即,那些不是非常重要的变异)。考虑到潜在的微生物遗传变异性,多重假设检验的程度非常高(高渗透率)将具有挑战性[65,66]。
对胃肠道以外的全身性疾病的研究也表明了特定肠道微生物菌株的功能作用。例如,在某些人群中,新发病的类风湿关节炎患者似乎在肠道中富含丰富的体育球菌(P. copri i),有证据表明该体育球菌亚群在功能或系统发育上可能是不同的[67]。肥胖和2型糖尿病(T2D)在整体肠道微生物组中显示出相对较弱的分类学或功能变化,但再次使用小鼠来避免人类种群结构的挑战,事实证明,粘蛋白Akkermansia muciniphila的特定菌株在减轻这些代谢条件方面起因[68] ]。在人类受试者中,至少有一项研究发现在T2D患者组中特异于拟杆菌的亚群具有SNP [69]。更广泛地说,在一项随机临床试验中,膳食纤维选择性地富集了几种SCFA生产者的菌株特异性促进作用,包括双歧杆菌,真细菌和乳杆菌,改善了T2D参数[70]。
桥接肠道微生物组,胃肠道和全身健康的最复杂条件之一就是癌症。 特别是在结直肠癌(CRC)中,特定的微生物菌株功能可以很容易地证明是局部原因,例如上述[71]或脆弱型芽孢杆菌毒素[72]引起的pks + +大肠杆菌DNA破坏大肠菌素的DNA。最近发现了其他微生物,例如核梭菌的CRC特定谱系,其机制包括Fap2介导与宿主Gal-GalNAc的结合[73]或通过TIGIT的免疫调节[74]介导了它们的致癌性和与典型口腔 F. nucleatum 菌株的区别。 。微生物对胃肠道或系统性癌症的其他影响机制仍未得到很好的理解,有强有力的证据证明了微生物对免疫疗法反应性的影响[75,76,77],但很少有菌株特异性的罪魁祸首。同样,有限的研究表明结肠内外的肿瘤内细菌能够直接代谢诸如吉西他滨等化学疗法[78],并有可能发现更多此类微生物-化学相互作用。
2.3 人体整个微生物组中的应变携带和变异 Strain carriage and variation in the body-wide human microbiome
虽然肠道微生物组的菌株流行病学可能是最发达的,但在整个人体栖息地中,“共生”和致病菌株的作用也存在类似的例子。与肠道一样,最极端的例子是经过充分研究的病原体[79],例如皮肤中的金黄色葡萄球菌的抗性变体和鼻微生物群[80]。最近,独立于培养物和基于高通量培养物的方法的组合暴露了数月至数年间受试者内病原体的进化[81]。在这些情况下,与pks + +大肠埃希氏菌一样,诸如mecA的抗性功能可以归因于一个或几个基因座,这些基因座通过可移动的染色体或质粒编码的元件在遗传上具有可变性[82]。然而,更出乎意料的是,最近的发现指出了与非病原体对应的菌株特异性相互作用,例如某些诱导葡萄球菌生物膜形成的角质杆菌(原丙酸杆菌)菌株产生卟啉III。实际上,由于其相对于肠道的生物地理异质性,暴露的地形表面(如皮肤,鼻咽和肺)是少数几个身体部位,可以直接观察到个体内多种竞争菌株的详细生态学和持久性[84,85] [86],例如,在牛皮癣中的表皮葡萄球菌菌株中[87]。
相反,在阴道微生物组中,个体内菌株的深分化在技术上更具挑战性。取而代之的是,这种环境揭示了优势乳杆菌和阴道其他物种之间的宿主之间存在广泛的亚种异质性,这再次引发了有关不同微生物进化枝中菌株和物种的确切定义的问题。具体而言,进化枝的系统对阴道优势乳杆菌(如詹氏乳杆菌,内氏乳杆菌,加氏乳杆菌和脆皮乳杆菌)的种内多样性进行分析非常复杂,该系统已根据分离株和分离株进行了重组研究。与培养无关的基因组学[88,89]。然而,随着时间的流逝,阴道乳酸杆菌和其他菌株在个体中可以保持相当稳定[90],特别大的环境变化,例如在怀孕过程中引起怀孕的转变[91]。就像肠道一样,菌株之间的这种遗传变异会影响健康,例如影响导致尿路感染的大肠杆菌的致病性[92,93]。在来自更急性传染病的例子中,由于微生物直接生化[94]及其对宿主免疫的影响[95],菌株特异性乳杆菌的生物活性本身可导致性传播感染如HIV的风险。
最后,口腔微生物学历史上提供了一些密切相关的微生物分离物之间表型异质性的第一个也是最惊人的例子[96,97,98],这种趋势在不依赖培养的测序和整个群落研究的时代依然存在。确实,一些最早的大规模微生物种群调查发现,口腔部位嗜性( oral site tropism)是亚种分化的强大驱动力[99,100,101],并且同一张嘴内不同表面(包括不同牙齿)的相关微生物定植者之间的遗传差异稳定。 。这些潜在的适应性强,利基特异性的变异体已开始大规模研究,在个体内长达数百天的个体内保持稳定[102],但揭示了进化枝成员如奈瑟氏球菌之间广泛的长期可塑性[11]。 。尽管关于口腔微生物生态学在牙周炎[103]到胰腺癌[104]和心脏病[105]中的作用的广泛研究正在进行中,但是口腔微生物群的生态学和基因组多样性已导致有限的菌株特异性迄今为止的关联。对于龋齿中的链球菌变异[106]或与口腔癌[107]相关的F. nucleatum,有人提出了一些建议,这暗示了其在CRC中的作用。其中包括足够的细节,以暗示微生物过程,例如多胺的生物合成,运动性和趋化性以及免疫刺激(例如LPS和鞭毛成分), 但是尚不清楚在复杂的口腔环境中这些功能可能分布的多种可能的菌株。
三、识别群落菌株多样性的策略和方法
我们的目标不是总结数十年来微生物学中用于区分培养物中微生物菌株的许多方法[108,109],因此我们将主要关注与培养无关的技术,以及一些方法基于高通量培养的方法适用于微生物群落(图1)。在这两种类别中,许多菌株定义方法都依赖于测序:基于培养物的分离物的组装,或基于扩增子的散弹枪宏基因组学或单细胞培养独立方法。其他分子测定法,尤其是基于质谱(MS)的蛋白质组学,可以应用于分离株或群落的菌株类型[110]。对于基于MS或NMR的代谢组学或代谢通量测量也是如此[111]。当然,自最早的微生物学以来,微生物培养生理学和直接成像已用于区分菌株,在某些情况下,这些经过时间考验的方法也可以应用于群落。
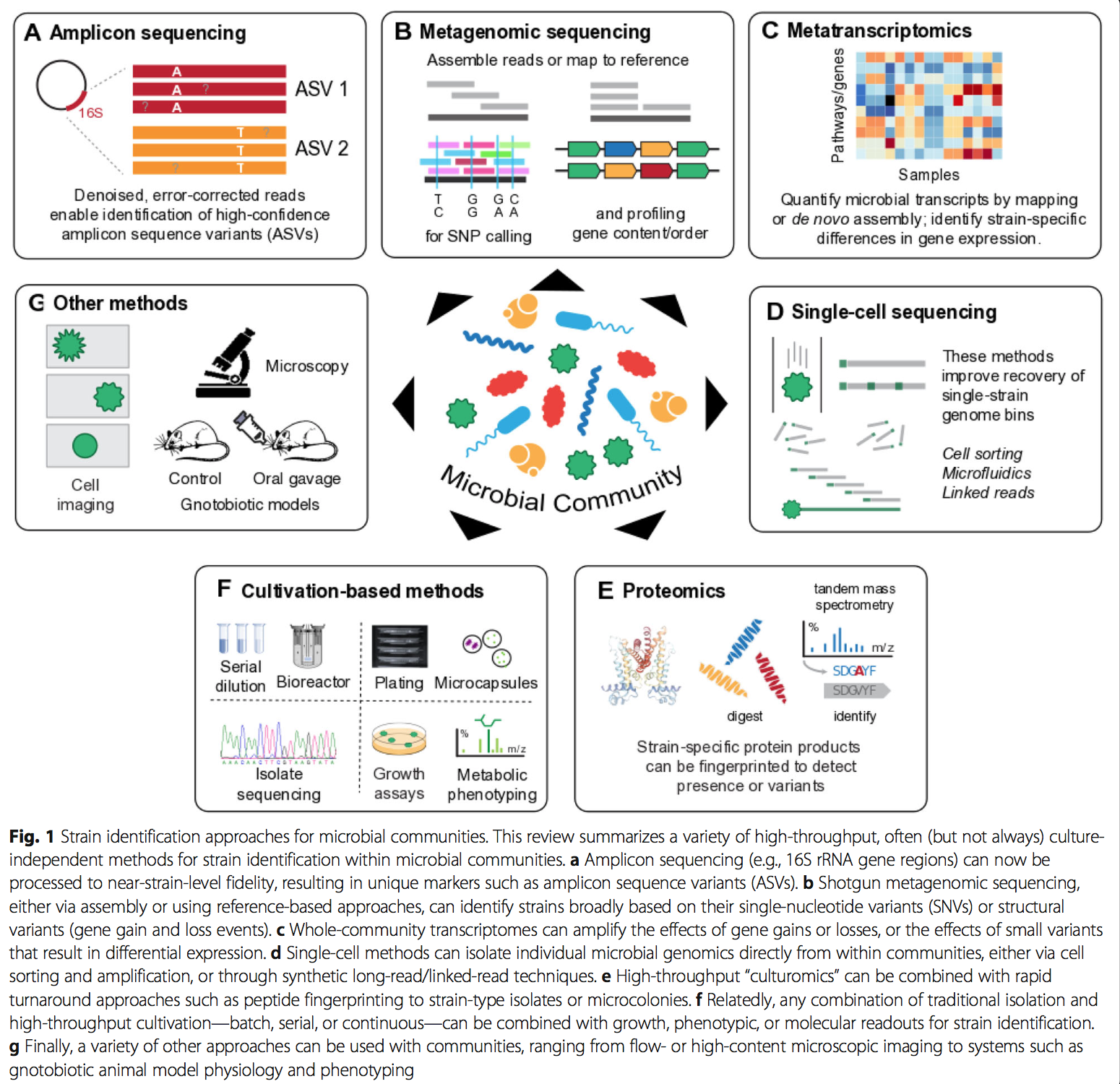
3.1 通过微生物群落测序鉴定菌株 Strain identification from microbial community sequencing
全群落测序在微生物菌株鉴定中的第一个突破,就像一般在群落范围内的第一个测序应用一样,来自依赖于扩增16S rRNA基因可变区的标记基因方法(扩增子或“ 16S”测序,表1)。在许多情况下,由于技术(测序误差,扩增噪声,生物信息学近似值)和生物学(扩增区域缺少核苷酸变异体)的局限性,基于扩增子的技术难以区分密切相关的微生物菌株[123,124]。然而,一旦数据生成平台达到了保留扩增子生物学变异所必需的保真度,那么就会出现几种计算方法,以可能的最菌株特异性方式对此类序列进行分类。寡聚分析[125,126]和最小熵分解[MED] [114]分别依赖于半监督和非监督分类,这些分类在其他方面相同的16S扩增子中的变异位置显示出不同样本之间的统计上不寻常的分布(因此不太可能是由于技术因素)。其他类型的子操作分类单位(OTU)聚类[113]随后使用统计错误建模(例如DADA2 [115])将这种直觉扩展到“精确”或“扩增子”序列变体(分别为ESV或ASV)或在序列同一性聚类之前或之后进行过滤(例如Deblur [116]或UNOISE2 [117])。菌株分离的16S扩增子已与此类方法一起使用,以非常特异地连接,例如将Porphyromonas asaccharolytica ATCC 25260和Parvimonas micra ATCC 33270连接至CRC,或评估肠道中菌株的时间稳定性[128]。 。通过额外的数据生成工作,它们通常还可以扩展到多个-[129]或非16S扩增子[130],例如用于识别用于扩增的最小目标基因座组的VaST系统[131]。尽管SNV多样性在基因组的子区域通常与整个基因组高度相关[8],但是在单个扩增的16S可变区内至少有一个可靠检测到的SNV的存在或不存在可以如此精确以至于成为高度进化枝-和特定于协议的[115]。

值得注意的是,全长16S rRNA基因测序的最早形式通过以高保真度捕获整个基因座的生物学变异而避免了许多这些问题[132], 随着“长读”技术的发展,最近在更高的吞吐量中再次成为现实。当前,三个主要平台可以提供此类long-reads:Pacific Biosciences,Oxford Nanopore , linked-read analogs,例如10X Genomics and Loop Genomics的产品。在这种情况下,可能最好地研究了太平洋生物科学循环共有测序(CCS)提供的极端保真度,可以轻松区分单核苷酸变体(SNV,尽管有时不是插入或缺失), 当它们在菌株之间的16S rRNA基因位点的任何地方存在时[133,134]。相反,虽然牛津纳米孔极具成本效益的MinION可以提供基本的全长16S rRNA基因读取,但其错误率将菌株特异性应用限制在群落中不存在与目标微生物高度同源的其他序列的情况下[135,136,137 ]。最后,现在存在几种协议,可在各种平台上促进“模拟”长读取或链接读取[138,139],但已达到商业可行性的那些尚待对微生物群落的扩增子谱进行正式评估[140]。类似地,这些技术有时可以应用于从单个细胞(例如,通过分选或微流体技术[48,141])或从交联的基因组拷贝[138]中分离的整个微生物基因组。如下所述,这消除了对真正的宏基因组学组装或分装的需求,尽管再次很少对这些新兴技术进行定量研究,以用于在菌株水平上进行全群落分析。
总体而言,shot弹枪宏基因组学方法可提供更丰富的微生物群落遗传组成图,因为它们原则上可以识别任何微生物基因组内任何地方的结构或SNV(表1)。 当前有两大类分析能够识别微生物菌株
- 第一类:基于宏基因组核苷酸(通常未组装)与参考基因或基因组的比对。 这通常是有效和敏感的,但当然只有在存在足够相似的参考基因组(或先前的宏基因组学组件[142,143,144])以允许直接绘制宏基因组读图时才有可能。 值得注意的是,“足够相似”的参考文献不必相对于目标元基因组具有特别高的识别性。 取而代之的是,它们必须简单地允许足够的全基因组作图,以鉴定出群落中毒株所特有的SNV或结构变异体,这可以在高达百分之几十的总核苷酸差异上获得成功。
广义地说,目前存在四类基于参考的群落菌株识别算法。
- 第一种方法使用一种基于歧义解析的阅读映射算法(例如PathoScope [118],Sigma [145])的量化方法,对与给定群落中最接近的一个或多个参考基因型进行识别。
- 第二个标识每个物种的显性,潜在的新基因型(菌株)。这些包括StrainPhlAn [8],MetaMLST [120],MetaSNV [146]和其他[37]。这些通常需要更深的测序(要靶向的菌株的覆盖范围达到10倍或更多),并且在选择要针对哪些参考序列进行选择方面存在差异(例如,完整的基因组与通用核心基因与物种特异性标记基因) SNV识别的方法和严格性。
- 第三类基于参考的方法将进一步尝试识别元基因组中每个物种的多个菌株,例如ConStrains [121]或DESMAN [122],它们需要更深的覆盖范围和更严格的噪声去除以防止假阳性。
- 最后,第四,依赖于结构变异而不是SNV变异的方法通常更敏感(适合群落成员,稀有度仅为〜1x或更低的覆盖率),并且包括PanPhlan [66](可以与基因靶向的功能分析器结合使用,例如例如HUMAnN [147]),MIDAS [37]和其他[4,65]。
或者,
- 第二类: 当没有足够相似的参考基因组时,宏基因组学组装[142,143,144]可用于高度新颖的菌株发现[148]。
在基于汇编的宏基因组菌株谱分析中存在固有的张力( inherent tension),因为大多数汇编程序都试图为每个重叠群识别单个共有序列,并且需要整个基因组(或区域)> 1×的覆盖范围。当单个菌株在一个群落中占据其附近的系统发育空间时,这是适当的,在这种情况下,可以通过将宏基因组读数映射回例如,一个装箱的装配体[149,150,151],并大致鉴定出完整基因组中的核苷酸或结构变体[8]。但是,在群落中存在太多紧密相关的菌株时,首先无法实现这样的共有序列,并且大多数组装者将无法提供适合作图的重叠群[152,153]。即使在可能的情况下,由于微生物群落装配体的高度生态和技术变异性,该过程可能会进一步复杂化,从而导致覆盖范围和置信度多样化(取决于测序深度和种群应变混合物,dependent on sequencing depth and population strain admixture),并受益于人工检测的推定变异[154,155 ]。促进这一过程的算法包括潜在应变分析(LSA),它可以使用跨多个相关(例如纵向)样本的协变聚类来优化应变级分类法[132]。同样,DESMAN使用的统计模型与扩增子数据中用于ASV调用的统计模型相同,以识别在多个样本的协同装配中得到良好支持的变异基因型[122]。迄今为止,在极少数情况下,已通过元转录组基因表达定量的类似差异,如心脏药物地高辛的埃格特氏菌新陈代谢的菌株特异性变异,确定了微生物群落中的菌株变异[49]。
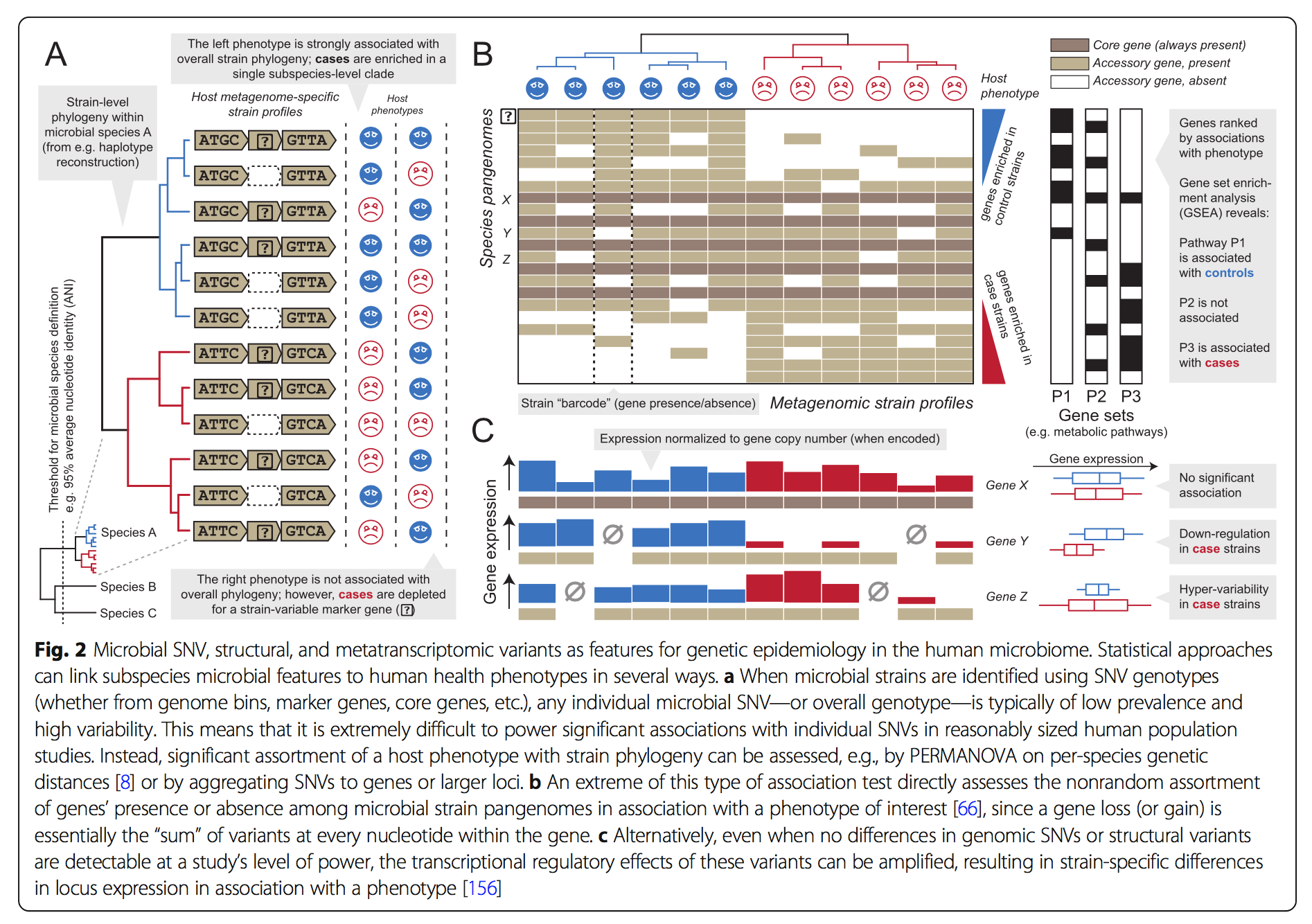
SNV与结构方法无论是来自参考序列还是装配体,通常都是互补的,并且可以提供有关同一基础群落的独特信息:SNV(在可检测时)可识别更细粒度的系统发育和进化差异,但在功能上难以解释,而结构变异(即,完整基因或基因组区域的获得或丢失)在群落内的检测下限较低,并且可以直接说明受影响基因的生化作用(已知,图2)。毫不奇怪,每种方法都可以提供不同的优点和缺点。结构变异可以通过基于参考的方法很好地捕获,该方法对独特的基因(非)检测很敏感。但是,使用这样的技术很难识别重排(而不是损益,rather than gains or losses),而可以通过基于装配的方法更好地识别它们(当它们可以与例如嵌合装配错误可靠地区分开时[157])。相反,SNV变异可通过基于参考或装配的方法很好地捕获(前者对具有代表性分离物的生物更敏感,后者对新生物则不那么敏感),并且可以通过全基因组或全基因组作图方法来捕获最独特的识别多态性发生的地方。最后,在微生物群落中,结构变异和较小程度上的核苷酸变异都受到遗传迁移机制的驱动,包括所有形式的侧向转移,基因增/减,移动元件,质粒和噬菌体整合。
3.2 在微生物群落中鉴定菌株的其他高通量分子方法
在群落中用于微生物菌株分型的其他分子技术通常仅限于可以培养或以其他方式分离的微生物,尽管(半)自动厌氧培养和纳米培养的进展也使高通量可行。特别是在临床微生物学中,通过质谱法肽指纹图谱进行近菌株变异分型对于病原体分离物[110、158]很常见,因为它的快速处理时间和相对于测序的每个样品成本低。该技术与上文介绍的从序列扩增子鉴定ASV有一些相同的警告(caveats),但是:
- 氨基酸变体必须存在于分析蛋白中的目标菌株之间,
- 且水平应高于实验噪声,
- 并且必须可分类为起源于参考数据库或通过聚类[159,160]。
虽然原则上可以使用MALDI-TOF MS技术在不依赖培养物的群落提取物中检测到相同类型的菌株水平蛋白变体,但此类应用仍然极具挑战性,相反,目前在基因组或分类中更普遍地分析群落蛋白质组学中心方式[161]。
相反,微生物成像(microbial imaging)(可以说是区分菌株的第一种方法)已使高通量以几种与培养无关的形式跃居整个群落,在适当的情况下,它们可以提供菌株水平的鉴定。在某些情况下,这可能意味着使用自动细胞分离和图像分析对微流体分离(或纳米培养)的细胞进行直接显微镜观察[162]。更多的分子技术包括光谱或组合荧光原位杂交(Combinatorial Labeling and Spectral Imaging或CLASI-FISH),该技术目前可以识别群落中的十几种微生物,同时保持空间结构[163,164]。连同相关技术,例如多标签FISH(MiL-FISH)[165],这依赖于在FISH探测的基因座(通常为16S rRNA基因区域)上存在足够的遗传变异,以通过光谱不同的探针进行差异性结合,但可以在某些情况下可扩展到活细菌[166]。对于其他微生物探针成像方法(例如流式细胞术[167]或光片显微镜[168])也是如此,它们可以保留活细胞,但需要具有能够区分密切相关菌株的基因座的探针或基因操作微生物。
尽管这些方法中有许多是部分或全部与培养无关的,但很难低估“culturomics”复兴在从包括人类微生物组在内的社区中分离和鉴定微生物菌株分离株的重要性[15、16、169]。虽然病原体流行病学长期以来一直依赖于多达数万种分离株中的比较基因组学,但直到最近才有效地从人群或个人中大规模分离出共生生物[170,171]。但是,这样做可以在不同空间位置[81、174]上的单个微生物组中,或随着时间的推移,在个体之间[12、13、172、173]中识别同一物种的分离株之间的菌株水平差异。 [170,175]。当然,一旦分离,这种微生物菌株就可以通过许多标准方法来表征,包括生长曲线或培养基之间的差异,化学(例如,抗微生物)抗性,代谢通量谱或扩增子或shot弹枪测序。或者,通过化粪池生物反应器(chemostat bioreactors)进行的全社区培养[176]提供了一个中间环境,在该环境中,原位稀有菌株可以席卷至优势地位,或以受控方式受到干扰,以扩增差异表型或序列,否则它们可能会保持在极限以下检测。最后,基于培养和不依赖培养的菌株鉴定技术在社区单细胞微生物分离[177,178]和微菌落生长[179,180]方面变得模糊。这种静脉中的微流体技术包括用于单细胞扩增的凝胶微滴(GMD)[181]或表型[182],以及将单个细胞的特异性与斑纹菌落的生物质相结合的微流片板(MSP)[183]。 (如果需要)。
特别是在考虑基于培养物的和离体/体外/模型系统分析时,与培养物无关的高通量流行病学与随后的菌株分离或操作相结合,为在人类微生物组中表征新型健康相关菌株提供了可能性。这篇综述从本质上讲是“自上而下”的观点,类似于正向遗传学,其中通过各种人群研究方法鉴定了感兴趣的菌株特异性特征(SNV,基因盒,新陈代谢等)[184]。通过从一级样品中分离[15,170]或从数据库或存储库(例如ATCC,BEI,DSMZ)对同源序列或相关菌株进行计算机检索,这种方法自然会导致这些变体的后续生化表征[ 185]。原始样品可通过gnotobiotics [186,187]或连续培养[188,189]或生长,表征或经过基因操作[15,190,191]的单个分离株表征为整个群落。此类方法与“自下而上”方法(类似于反向遗传学)很好地吻合,后者通过直接从分离株开始并评估其在gnotobiotic单或组合定植中的表型[192,193,194,195,196,197]或在可能的情况下,通过人工喂养来鉴定和表征与健康相关的菌株[198,199,200]或微生物群移植的临床试验[201,202,203,204,205]。
四、前景和未来方向
如上文所述,“菌株”(strain)的精确定义在整个生物学中有些不确定,更不用说微生物学[3]或微生物群落生物学[206]。尽管过去最常提到单一菌落分离培养物,但在微生物群落中引入可精确解析的遗传变异鉴定的技术和工具,导致该术语的拓宽。现在它以某种频率使用,表示具有相对较低遗传多样性的亚种或种内进化枝,其由核心或基因组同一性,扩增子内的核苷酸同一性(例如16S rRNA基因)或上述其他基因分型或表型相似性定义。正如有关微生物系统整体的文献中越来越多地讨论过的那样[8,207],这表明需要对菌株或亚种进化枝,尤其是在自然变异的微生物群落中进行更定量的定义。在没有单一共识定义的情况下,对于单个研究在描述基于培养或(特别是)与培养无关的微生物群落研究时,预先定义其“菌株”的用途非常有用[174]。
不管其精确定义如何,几种新兴技术提供了令人兴奋的新方法来鉴定,分离和表征人类微生物组中与健康相关的菌株。从历史上看,与明显的急性表型无关的微生物遗传变异在很大程度上未被发现,直到可以有效地捕获它们的全社区分析技术的相对较新的可用性为止。迄今为止,真正依赖于单个微生物分离的单细胞方法很难应用于人类流行病学,真核细胞的方法不能很好地大规模转移到微生物细胞壁生物化学的异质性[208],而环境群落特征分析的方法很难适用于人类粪便或皮肤等各种基质[209]。除了用于细胞分离和裂解的生物工程外,低输入,低噪声的DNA分离,扩增和测序方面的进展将有助于应对这一挑战[210],而内在扩增基因组的纳米培养方法也将有助于解决这一难题[180]。此类从人类微生物组捕获菌株的方法与其他用于大规模表征其特征的技术并驾齐驱,包括更便宜的实验系统,例如芯片上的肠道[211,212]或类器官变体[213,214]在单一分离培养物和丰富的gnotobiotic模型之间。最终,了解人类微生物组生物学不仅需要检测社区中特定的微生物遗传变异,还需要对其进行引入和操作,包括从宿主社区中分离后甚至在遗传上对任何微生物菌株进行遗传扰动的理论能力[173,190 ]。
即使在没有这种技术的情况下,仍然需要做大量工作来表征已经发现的人类微生物组中的微生物菌株多样性。在人类微生物组中鉴定的数千万个基因家族中[23,99,215],约75%的生物化学特征与参考序列的同源性(在某些情况下较远)仅是(在某些情况下是远程)同源性,而约25%的基因家族没有与任何孤立的开放阅读框架非常相似[216]。这种惊人的生化暗物质池对微生物生物勘探者来说并不奇怪,他们数十年来主要为新的酶和抗菌功能而开采环境群落[217]。因此,它也代表着在人类健康中发现新的生物活性物质的巨大潜力,因为与人类相关的微生物很容易富含调节宿主反应的蛋白质和代谢产物[218]。在上述许多示例中,微生物组中SNV或结构变异与人类表型或环境因素的成功关联已导致功能未知的基因[13,65,66]。因此,人类微生物组中的菌株级流行病学可以帮助确定和鉴定与健康最相关的“最有趣的”新型微生物变体和产品这一艰巨的任务。
最后,微生物组中更好的菌株表征技术可以有益于人类健康的方式本身是多种多样的。廉价,快速且可重现的方法可量化人群中微生物组SNV和遗传变异,这将有助于鉴定精确的微生物危险因素,就像全基因组关联研究(GWAS)的人类遗传学平台标准化一样[219]。同样类似于GWAS,微生物菌株可因此提供疾病风险或诊断的预后或诊断生物标志物,或暗示其潜在分子机制[220,221,222]。对于比较遗传学的微生物分离株,这种情况已经存在了数十年,并且随着元基因组的数量和深度的不断增加,它无疑也将在微生物群落中变得可行[223,224]。相反,发现具有生物活性的菌株的特征可用于开发新的干预措施以维持健康或进行治疗。这些可以包括基于供体或受体菌株含量更好地针对现有粪便微生物菌群移植(FMT)技术[225],合成FMT的合理设计[226],FMT或益生元的治疗反应预测[227,228,229,230]或最终管理转基因生物或社区[231,232,233,234]。因此,最近在微生物群落和人类微生物组的菌株级流行病学方面的工作是实现微生物组巨大的翻译潜力的许多重要且正在进行的工作之一。
缩略语
- ASV: Amplicon sequence variant
- CCS: Circular consensus sequencing
- CLASI-FISH: Combinatorial Labeling and Spectral Imaging
- CRC: Colorectal cancer
- EHEC: Enterohemorrhagic E. coli
- ESV: Exact sequence variant
- FMT: Fecal microbiota transplant
- GI: Gastrointestinal
- GMDs: Gel microdroplets
- GWAS: Genome-wide association studies
- HMO: Human milk oligosaccharide
- IBD: Inflammatory bowel diseases
- LPS: Lipopolysaccharides
- MED: Minimum Entropy Decomposition
- MS: Mass spectrometry
- MSPs: Microfluidic streak plates
- OTU: Operational taxonomic unit
- SCFA: Short-chain fatty acid
- SNP: Single nucleotide polymorphism
- SNV: Single-nucleotide variant
- T2D: Type 2 diabetes
参考资料
- Yan, Y., Nguyen, L.H., Franzosa, E.A. et al. Strain-level epidemiology of microbial communities and the human microbiome. Genome Med 12, 71 (2020). https://doi.org/10.1186/s13073-020-00765-y。 地址: https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-020-00765-y
