【2.5】蛋白质序列空间(protein sequence space)
In evolutionary biology, sequence space is a way of representing all possible sequences (for a protein, gene or genome). The sequence space has one dimension per amino acid or nucleotide in the sequence leading to highly dimensional spaces.

对于100个氨基酸的蛋白质,序列空间大小的典型估计是20^100(约10^130)
摘要
我们建议,通过考虑生物数量的上限和下限,基因组大小,突变率和功能不同的氨基酸类别的数量,在生命的地球填充期间,蛋白质序列空间的广阔性实际上是完全可探索的。 我们得出的结论是,在最后4 Gyr中,生命只探索了序列空间的一小部分,而对所有功能性蛋白质序列空间进行探索是相当合理的,而且在分子水平上,没有 应急的作用。
注:天文学家使用Gyr或Gy作为gigayear(10^9 years) 的缩写
一、前言
在考虑地球上生命历史中功能蛋白的分子进化时,通常会做出两个假设:
- 首先,蛋白质序列空间( protein sequence space)的大小,即可能的氨基酸序列的数量,在天文学上是巨大的;
- 其次,在地球上的生命过程中仅探索了无限小的部分(例如Salisbury 1969; Maynard Smith 1970; Mandecki 1998; Luisi 2003; Carrier 2004; de Duve 2005)。
Luisi和Chiarabelli将序列空间的未开发部分称为含有“从未出生的蛋白质”(never born proteins)(Luisi等人2006; Chiarabelli&de Lucrezia 2007)。我们希望通过估算自4 Gyr之前的生命起源以来可以探索多少这个空间来讨论这两个假设。如下所述,其他人得出的结论是,第一个假设是不正确的,我们同意这个结论。然而,我们还得出结论,第二个假设是不正确的,并计算出大部分序列空间可能已被探索过。
如何定义这个 protein sequence space呢?
在讨论第二个假设之前,我们希望总结显示第一个假设的信息,即序列空间很大,是假的。对于100个氨基酸的蛋白质,序列空间大小的典型估计是20^100(约10^130),其中可以发现任何正常发生的20个氨基酸。这个数字确实很大,但很可能是对蛋白质序列空间大小的高估。例如,Dill及其同事使用简单的理论模型来建议(Lau&Dill 1990; Chan&Dill 1991; Dill 1999),并且蛋白质序列的实验或计算变异提供了充分的证据,蛋白质中大多数氨基酸的实际身份是无关紧要的。自然界中的一个实例可以是原核DNA甲基转移酶,其各自含有约150个氨基酸的靶识别结构域(target recognition domain,TRD),其识别通常长度为3-6bp的特定DNA序列,和保守的催化结构域。数千种已知的TRD序列显示出可忽略的氨基酸序列保守性,尽管它们需要识别的核苷酸序列数量相当有限。
作为减少序列空间大小的极端方法,Dill(1999)提出只需要两种类型的氨基酸来形成亲水和疏水的蛋白质结构,而且仅定义至关重要的蛋白质表面。 这两个建议分别将序列空间的大小减小到2^100和2^33(即大约10^30和大约10^10)。值得注意的是,最近的粗粒度“管”模型(coarse-grained ‘tube’ models)甚至更进一步并且去除了所有原子信息,仅留下了与tube的其他部分相互作用的潜在能量函数。尽管该模型具有极度粗糙的粒度,但仍然可以找到可识别的“蛋白质”结构(Banavar等人,2006)。
虽然这似乎违背了Anfinsen的教条,即蛋白质结构由其氨基酸序列决定(Anfinsen 1973),但它实际上只是氨基酸’字母’大小极度减少的情况。获得的管结构非常类似于自最后一个通用祖先以来显然保守的序列所采用的短折叠区段(Sobolevsky&Trifonov 2006)。当已知许多蛋白质是模块化的并且含有少至约50个氨基酸的结构域从而将空间减少到20^50(或大约10^65)时,假设蛋白质链长度必须至少为100个氨基酸也会增加序列空间的大小。所有这些粗粒化方法的结论是,减少的氨基酸字母表能够产生所有蛋白质折叠(大约几千个离散折叠; Denton 2008)并提供能够支持所有蛋白质功能的支架(我们对于目前的讨论,我们会忽略原生未折叠蛋白质的空间,但由于这些蛋白质通常在执行其功能时会折叠,因此这种区别对我们的目的并不重要; Dyson&Wright 2005)。功能的相空间可能比折叠空间的大小大一些数量级,因为宏基因组学项目揭示了由新蛋白质序列的数量决定的越来越多的未知蛋白质家族(Raes等人,2007)。然而,目前尚不清楚新的折叠是否存在,因为保守的折叠,例如TIM tube,能够显示许多功能(Nagano等人,2002)。
为了进一步支持减少氨基酸字母表的这一想法,还有一些非常合理的建议,即原始氨基酸库(original amino acid repertoire)只有四到五个氨基酸,如Miller-Urey实验和Murchison meteorite 中发现的那些,遗传密码最初仅限于目前仍在蛋白质中占主导地位的少数氨基酸。 具有减少的氨基酸库( amino acid repertoires )的蛋白质可以成功折叠和起作用。
图1显示了作为不同氨基酸(或氨基酸类别,1-20)的数量和功能上重要的氨基酸链(33,50或100)的长度的函数的可能序列的数目。 它突出了序列空间大小的急剧减少,如果将可用氨基酸类型的数量限制为小于目前通常发现的20种,这种限制似乎在实验上是合理的。
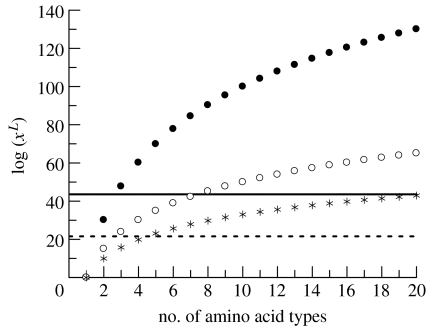
图1
蛋白质序列空间log(x^L)的大小作为含有33个(星号),50个(空心圆圈)或100个(填充圆圈)氨基酸(长度L)蛋白质的氨基酸字母表大小(即不同类型氨基酸的数量,x)的函数。水平线代表自地球上生命起源以来4 Gyr期间探索的最大(实线)和最小(虚线)序列的估计值。
二、结果
我们现在希望考虑关于蛋白质序列空间的第二个假设:地球上的生命只探索了一个无穷小的部分。为了研究可以探索多少序列空间,最简单的做法是使用一些自由假设对生命起源以来产生的独特氨基酸序列的数量进行上限和下限估计。考虑到上限,很明显细菌在细胞数量的乘积(10^30; Whitman等人1998)乘以每个基因组中的基因数量(10^4,一个小的高估)中占主导地球。让我们假设这10^34的总基因中的每一个基因都是独特的,并且进化已经在4 Gyr的这些基因上工作,每一年将每个基因完全改变为其他一些独特的新基因。这给出了自生命起源以来探索的4×10^43种不同氨基酸序列的极限上限。病毒和真核基因组对这些序列的贡献很难估计,但它不太可能比细菌的4×10^43序列大几个数量级。如果它们的贡献相似或更小,则可以在粗略计算中忽略它。为了与我们的计算进行比较,Mandecki(1998)给出了自生命起源以来10^50个蛋白质序列的限制。探测序列数量的下限更难以估计,但据估计地球上有109种不同的细菌物种(Whitman等人1998; Medini等人2005; Simonson等人2005)。如果我们假设每个物种都有10^3个序列的独特互补(低估),并且每个物种每个物种只有一个序列发生了变化(基于细菌突变率分析的合理估计; Perfeito et al.2007),生成时间为1年(对于许多现代细菌而言相当低估(Ochman等人,1999),但对于古代有机体或在贫瘠环境中缓慢生长的生物可能是合理的),那么我们得到的数字为4×10^21不同自生命起源以来测试的蛋白质序列。
这两个限制如图1所示。尽管经常引用的20^100(约10^130)序列空间大小远远超过这些限制,但对序列空间大小的其他更可信的估计,特别是有限的氨基酸多样性或减少长度,接近或在这两个限度内。考虑到上限,如果链长度分别为33,50和100个氨基酸,则已经探索了含有20,8和3种氨基酸的所有序列。考虑到下限,实际上可以探索分别含有五种或三种氨基酸的长度为33和50个氨基酸的所有链。 (对只有两种残留物的100个氨基酸的较长链的探索显然不那么完整,但它不是总量中可忽略的一部分。)因此,对于所有实际(即功能和结构)目的而言,完全可行的,蛋白质序列空间在地球生命进化过程中(甚至在真核生物出现之前)得到了充分的探索。
三、结论
蛋白质序列空间通常被视为失调序列的无限沙漠,只有少数绿洲的工作序列通过狭窄的途径连接(Ax 2000,2004)。通过自然选择在该空间上的导航是困难的并且可能采取许多不同的途径,因此导致具有大不相同的蛋白质组成的生物。这种偶然性的想法,如果在物种层面上采取,导致古尔德建议(Gould),如果要重新运行“生命的磁带”,那么进化将采取完全不同的路径,我们作为一个物种,只会出现高度不可能的事故(Gould 1991; Luisi 2003; de Duve 2007a,b)。然而,如果我们的简单计算有任何优点,那么蛋白质序列分析不能在分子水平上对偶然性的概念提供支持,并且它为收敛的思想提供了强有力的支持(Conway Morris 2000,2004; Dawkins 2005; Vermeij 2006; de Duve 2007a,b)。如果要重新运行磁带,那么生物体的蛋白质组成将是相似的。我们的计算消除了对自然选择的几乎不可能的不切实际的压力,以便在蛋白质序列空间中导航,通过简单地指示大多数序列已被尝试在某种程度上有用而避免大量无功能序列,并且有许多可能的途径来获得蛋白质理想的功能(Nagano等,2002; Anantharaman等,2003; Holliday等,2007)。
最后,我们得出结论,20^100年和类似的大数字仅仅是“稻草人”(‘straw men’ ),以与 蛋白质折叠率的’Levinthal悖论'(Levinthal 1969; Zwanzig等1992)。 20^100现在没有比博尔赫斯(1999)梦幻般的“巴别图书馆”( Borges' fantastical ‘Library of Babel’ )中出现的大约2×10^1834097本书更有用,并且与氨基酸和蛋白质的真实世界没有关系。 因此,我们希望我们的计算也将排除任何可能使用这个大数字’游戏',来为假定神圣干预提供理由(Bradley 2004; Dembski 2004)。
参考资料
- J R Soc Interface. 2008 Aug 6; 5(25): 953–956. How much of protein sequence space has been explored by life on Earth?
- https://en.wikipedia.org/wiki/Sequence_space_(evolution)
