【3.4.2】三级结构的获得与预测
一、实验方法获得三级结构

- X射线衍射法: 必须先得到蛋白质的晶体(X- ray crystal diffraction: target proteins have to be crystallized first)
- 冷冻电子显微技术(Cryo-electron microscopy or CryoEM)(不需要固定,可以有多个结构,这也就是有多个model的原因?)
- 核磁共振法: 不能大于120aa (NMR, less than 120aa)
实验的缺点:
- 实验材料要求高
- 实验仪器造价高
- 实验耗时成本高
计算方法预测三级结构:
- 同源建模法 homolog modeling
- 综合法 ensemble method
- 穿线法 threading
- 从头计算法 ab initio
- 模型质量评估
二、三级结构预测
2.1 同源建模法 homolog modeling
2.1.1 原理
相似的氨基酸序列对应着相似的蛋白质结构
2.1.2.步骤流程
- 找到与目标序列同源的已知结构作为模板(目标序列与模板序列的一致度要 ≥ 30%)
- 为目标序列与模板序列(可以多条)创建序列比对。通常比对软件自动创建的序列比对还需要进一步人工矫正。
- 根据第二部创建的序列比对,用同源建模软件预测结构模型。
- 评估模型质量,并根据苹果结果重复以上过程,直至模型质量合格。
2.1.3.工具介绍( SWISS-MODEL )
SWISS-MODEL是一款用同源建模法预测蛋白质三级结构的全自动在线软件。
SWISS-MODEL中一共有三个工作方式:
- First Approach mode:
- Alignment Interface mode:
- Project(Optimise)mode:
SWISS-MODEL还能对寡聚蛋白质和GPCR(G Protein-Coupled Receptors )进行单独的建模
PS: 30%的相似度这个标准怎么来的?
2.1.4. 预测效果(使用范围)
如果目标序列与模板序列一致度极高,那么同源建模法是最准确的方法。
- 如果一致度能达到30%,那么模型的准确度就可以达到80%,模型可以用于寻找功能位点,以及推测功能关系等。
- 如果一致度能达到50%,那么模型的准确度就可以达到95%, 可以根据模型设计定点突变实验,设计晶体结构自转,辅助完成真是结构的测定
- 如果一致度能达到70%以上,我们可以认为预测模型完全代表真是结果,可以用来分子筛选,分子对接,药物设计结构功能研究。
特殊情况,虽然序列一致度达到很高水平,但是结构却并不相同。(这种情况比较少见,但需要注意)。 同时,此方法适用于能找到相似度高的已知结构的序列 。
2.2 穿线法 threading
2.2.1 原理
- 不相似的氨基酸序列也可以对应着相似的蛋白质结构。
- 已知的蛋白质结构约10万个,其所具有的不同的结构拓扑只有1393个,且自2008年就没再有新的结构拓扑产生。
- 将不同的序列放在已知的拓扑结构中,看谁的能量最低,最低的那个即为可能的模型。
Threading方法不通过序列相似性比较来判断两个蛋白质的结构是否相关,而是直接判断待测序列和已知结构模板间的相关程度。Threading方法认为天然结构中残基间相互的吸引或者排斥有一定的倾向性,也就是说某些残基出现在一定空间范围内对结构有稳定作用,而另外一些则会使结构变的不稳定,并假设这种作用能有一个能量函数加以描述。计算某条蛋白质序列安放到结构模板之后其残基间这种作用力分值,通过结果来判断未知结构和该模板结构之间的相似性。能量函数是通过统计已知结构库中残基对在一定范围内出现的频率,这个频率反映了残基间吸引或者排斥倾向,这个频率转换的分值也通常称作接触能。
2.2.2 折叠识别的网络服务
- FUGE: http://www-cryst.bioc.cam.ac.uk/~fugue/
- 3D-PSSM:http://www.sbg.bio.ic.ac.uk/~3dpssm/index2.html
- Gen-THREADER(next)
- GenTHREADER http://bioinf.cs.ucl.ac.uk/threader/ (下载)http://bioinf.cs.ucl.ac.uk/psipred/psiform.html (PSIPRED 在线服务的项目之一)
- Gen-THREADER应用二级神经网络在折叠模板库中搜索目标蛋白质的结构模板。用户只需要提交目标蛋白质的序列,Gen-THREADER就会通过邮件返回预测的结果。邮件返回的结果中将包含排列在前面10位的折叠模板的名称、预测可信度(分为Certain, High, Medium, Low, Guess五个不同的等级)、比对能量值以及目标蛋白质和模板蛋白质的序列比对结果。(next)
2.2.3 预测工具:I-TASSER
I-TASSER是一款用穿线法预测蛋白质三级结构的在线软件,在连续几届蛋白质结构预测比赛中皆排名第一。作者为美国密歇根大学的张阳教授。
CASP- Critical Assessment of protein Structure Prediction 蛋白质结构生物信息预测国际竞赛。主要目标是对当前在蛋白质结构预测领域的“能为”和“不能为”作一深入且客观的评价。为实现这一目标,参与者们对一组即将公开的结构进行预测,并根据预测准确度排名。上海交通大学电子信息与电器工程学院的沈宏斌教授研究组在CASP11中取得第三名。
I-TASSER 一个用户一次只能提交一个任务,一个IP地址只能提交一个任务。
近40个小时,I-TASSER可以预测出结果。
I-TASSER预测的五个模型,包括:
- 模型质量评估系数 C-score:[-5,2],分值越高模型可信度越高;
- TM-score: 两两结构相似度系数, >0.5说明模型具有正确的结构拓扑,可信;<0.17 说明模型属于随机模型,不可信,
- RMSD:两两结构件的距离偏差。
PDB数据库中,与排名第一的预测模型从结构水平上最相似的结构。模型结构以卡通显示,PDB结构以细管显示。排名第一的预测模型所涉及的TM-score 和RMSD即由词表算出。
2.3 从头计算法 ab initio
2.3.1 原理
1973年《科学》 Anfinsen:蛋白质的三维结构决定于自身的氨基酸序列,并且处于最低的自由能状态。
Anfinsen于1974提出蛋白质天然构象是处于全局自由能最小状态,这就为通过计算蛋白质构象能来预测蛋白质三级结构提供了理论依据。
从头预测方法存在两个方面的问题:
- 首先,蛋白质折叠过程是一个非常复杂的动力学过程,受蛋白质组成以及外界(溶液)环境的影响,如今还没有一个很好的理论能描述这个过程。
- 其次,从头预测方法将自由能最小的构象作为天然的构象,能否找到这个天然构象还取决于选取的能量函数是否能真实的反映蛋白质内部分子间相互作用以及能量关系,还没有一个很好的能量函数能反映蛋白质折叠。
正是由于这些制约,相比较前面所讲的同源建模、二级结构预测和折叠识别等方法,从头预测的方法目前并没有得到大范围的应用。
2.3.2 工具
QUARK是一款从头九三法预测蛋白质三级结构的在线软件,适用于没有同源模板的蛋白质,且氨基酸序列长度200以内。
2.4 综合法
2.4.1 原理
综合了同源建模法、穿线法和从头计算法等多种方法,将氨基酸序列分段,情况不同的片段采用不同的方法。
2.4.2 工具
ROBETTA综合了同源建模法和从头计算法两种方法。能找到模板的区域用同源建模法,找不到的区域用从头计算法。
整条序列被分成多个domains,每个domain采用不同的方法分别预测。同源建模法需要几个小时到几天的时间,从头计算法需要几天到几周的时间,这取决于目标序列的预测“难易”程度。
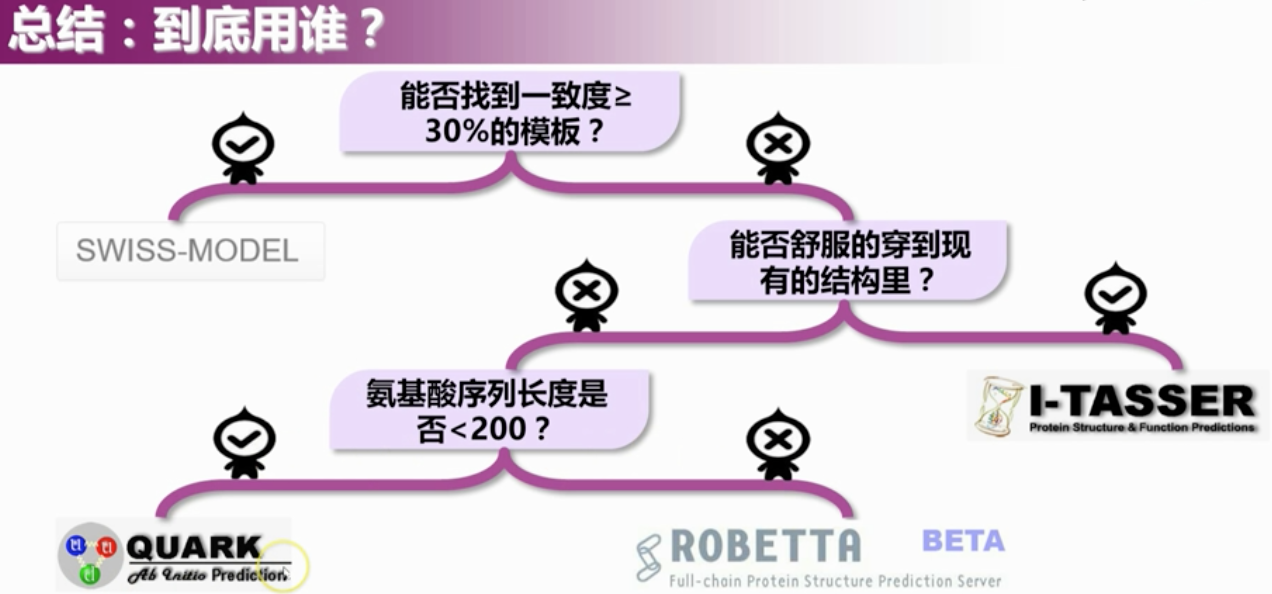
三、总结
3.1 比较
3.2 模型质量评估(Model Quality Assessment programs,MQAPs)
对于通过计算方法获得的模型,必须进行必要的模型质量评估,以确定模型的可靠性。模型质量评估软件并不比较预测模型跟真事结构的差别大小,而是从空间几何学,立体化学和能量分布三方面评估一个模型的自身合理性。
例如:
- I-TASSER: C-score的取值范围是[-5,2],分值越高模型越可靠。
- Swiss-Model: QMEAN4的取值范围是[0,1],分值越高模型越可靠。
- QUARK :TM-score >0.5 说明模型的可靠,<0.17 说明模型不可信。
- SAVES提供6个模型质量评估软件。可以一次6个软件一起作,也可以单独选择 Verify 3D、 PROCHECK 和 ERRAT 。
- Verify 3D : 超过80%的残基拥有大于0.2的3D/1D值,则模型质量合格。 低质量部分,需要进一步修正。
- PROCHECK:拉氏图检查Cα的两面角()是否合理。合理的模型超过90%的残疾都应该位于红色的容许区域和正黄色的额外容许区域。落到其他区域的残基应当被查看并修正。
- ProQ: 通过LGscore和MaxSub两个值反应模型质量。计算时间只有十几秒,并且评估结果明确。
- ModFold评估结果明确。评估需要30分钟左右,并且一个email地址一次只能提交一个评估任务。
3.3 蛋白质结构预测的策略
第一步:判断目标序列中是否包含关键性的特征:
- 跨膜片段
- 查寻这个蛋白质中可能存在的已知结构域,如用 Interpro、PSI-BLAST之类的工具
第二步: 是否能采用比较建模法
- 当不能用比较建模时,下一步则应该是二级结构预测
- 对于球蛋白的结构域的预测要比膜蛋白更加准确
- 二级结构预测完成之后则是进行折叠识别
- 预测精度通常也要比标准比较建模法低得多
蛋白质结构预测技术评估大赛 (Critical Assessment of Techniques for Protein Structure Prediction,CASP): http://predictioncenter.org/
CASP是一个世界性的蛋白质结构预测技术评比活动。1994年,第一届CASP在美国马里兰大学生物技术研究所的约翰·莫尔特(John Moult)倡议、组织下举行,此后每两年举行一次。
First Approach mode:
如果没有除序列之外的任何信息,那么可以首先用First Approach mode来决定序列是否能通过同源方法建模。直接提交序列,SWISS-MODEL将在已知结构蛋白质数据库中搜索它的同源蛋白质,只有当序列相似度大于25%时才会建立结构模型,并返回寻找到的模板结构。
Alignment Interface mode:
如果已经得到了这条蛋白质的同源蛋白质以及它们的多序列比对结果,而且它的同源蛋白中包含了已知结构的蛋白质,那么可以通过Alignment Interface mode直接进行结构建模。
Project(Optimise)mode:
对First Approach mode得到的结构模型进行优化,Project mode利用生物化学信息来修正结构模型上存在的能量不合理区域。并且Project mode允许用户自行调整,以得到更精确的模型。
参考资料
- 山东大学 生物信息学课题组荣誉出品 http://www.crc.sdu.edu.cn/bioinfo 巩晶老师课件
- 李裕强老师的课件 http://wenku.baidu.com/link?url=942lI1TEoY6tmC4T2RHZIGqqCWPqHoYMN3cOVUdKN9a7z7ce2FoYwYllHPwieBv51foAEu1qjaFmYDLES9CzFbV1Pg4V4wyW8bDJzknNHty
- 南京大学 杨荣武老师 《结构生物学》课件
