【6.1.2】CRISPR-Cas9 gRNA设计工具的比较
CRISPR sgRNA预测常用软件
- E-CRISP http://www.e-crisp.org/E-CRISP/
- ChopChop http://chopchop.cbu.uib.no/
- CRISPOR http://crispor.tefor.net/
- Broad Institute GPP https://portals.broadinstitute.org/gppx/crispick/public
- Benchling https://www.benchling.com/crispr/
基于CRISPR的基因编辑的流行导致产生了大量用于设计CRISPR-Cas9指南的工具。这也受到以下事实的驱动:在使用CRISPR进行基因编辑时,设计高度特异性和有效的指南是一项至关重要的任务,而不是琐碎的任务(but not trivial)。在这里,我们彻底分析了18种设计工具的性能。根据运行时性能,计算要求和生成的指南对它们进行评估。为实现此目的,我们实现了一种在执行给定工具时审核系统资源的方法,并在源自鼠基因组的大小递增的数据集上测试了每个工具。我们发现只有五个工具具有计算性能,这使它们可以在合理的时间内分析整个基因组,而不会耗尽计算资源。所确定的指南存在很大差异,一些工具报告了所有可能的指南,而其他工具则进行了过滤以提高预测效率。一些工具也未能排除针对基因组中多个位置的指导。我们还考虑了两个集合,每个集合都有一千多个指南,可提供实验数据。数据集之间的性能差异很大,但是工具的相对顺序是部分保留的。重要的是,最惊人的结果是工具之间缺乏共识。我们的结果表明,CRISPR-Cas9指导设计工具需要进一步的工作才能实现快速的全基因组分析,并且指导设计的改进可能需要结合多种方法。
作者总结
现代基因组工程技术提供了前所未有的DNA修饰方法。基于CRISPR的系统由于其有效性和相对简单性,引起了大家的高度关注。但是,针对特定区域使用的指南的设计并非易事。研究人员既需要使进行所需修改的可能性最大化,又要使不希望的偏离目标的风险最小化。为了帮助研究人员做出更明智的决策,已经开发了许多软件工具来辅助指导设计。在这里,我们分析了CRISPR-Cas9指南的18种设计工具的性能。我们根据工具的运行时,计算要求以及它们生成的输出来评估每个工具。我们对规模不断扩大的数据集(以评估其可扩展性)以及可获得实验数据的指南进行了基准测试。我们的结果表明,工具之间几乎没有共识,指南设计的改进可能需要结合多种方法。
指南 (guide) 代表引物
一、前言
在古细菌和细菌中发现了野生型CRISPR(聚簇的规则间隔的短回文重复序列),可作为一种适应性免疫系统[1]。 CRISPR能够通过三个步骤[2]提供免疫方法:
1.从入侵的噬菌体中获得DNA片段,并将其存储在CRISPR阵列中,以记忆过去的病毒感染; 2. CRISPR区表达并成熟以产生先前获得的DNA片段(或指南)的重复片段; 3. 对于化脓链球菌Cas9(SpCas9),由于向导DNA的同源性,向导与SpCas9核酸酶结合以实现位点特异性切割。最后一步提供了对宿主细胞的免疫力,也是CRISPR在提供合成指南的基因组工程环境中使用的机制。
基于CRISPR的系统已用于许多此类应用[3-5]。但是,指南(guide)的设计并非易事,因为指南的效率和特异性是至关重要的因素。
因此,采用计算技术来识别和评估候选CRISPR-Cas9指南。 在这里,我们分析了18种CRISPR-Cas9指南设计方法,以评估它们是否适合用于快速全基因组分析,以及潜在的组合方法是否可以实现质量更高的解决方案。可用的工具可以基于算法方法进行分类(即程序或通过使用实验数据训练的模型);但是,在构建工具集时,我们考虑了多种因素:
- 是否容易获得源代码,
- 安装过程是否简单明了,
- 该工具仅不提供用于执行以下操作的包装器正则表达式和(iv)指南长度和PAM序列可以自定义。
在我们的分析中,我们不仅要考虑它们的输出(即确定的目标),还要考虑它们在合理时间内加工整个基因组的能力。这对于大型基因组(例如某些开花植物)尤其重要。对于某些应用程序(例如研究需要靶向多个基因的复杂途径或功能)(例如睡眠[6-8])或生成全基因组图谱,这也是至关重要的功能
二、工具总结 review
我们选择了18种在开放源代码许可下发布的指南设计工具,并报告了化脓性链球菌Cas9(SpCas9)核酸酶的候选指南;表1中列出了这些工具。每个工具都使用了最新版本,有关详细信息,请参见补充数据。
Python(n = 11)和Perl(n = 5)是最常见的编程语言,CT-Finder和CRISPOR也通过PHP(由JavaScript,CSS,HTML等补充)实现基于Web的界面。为了缩短运行时间,mm10db在C中实现了其某些组件。SSC和FlashFry分别在C和Java中实现。 PhytoCRISP-Ex是Perl实现的工具,但是,它广泛使用Linux bash命令进行预处理。工具的配置通常是通过命令标记实现的,但是,诸如Cas-Designer和CasFinder之类的工具是通过文本文件配置的。 CHOPCHOP通过主脚本文件中的全局变量进行配置。只有Cas-Designer可以利用GPU来获取其他计算资源。考虑到计算性能,我们在CPU和GPU模式下都运行了该工具。
SciPy [10](包括Numpy)和BioPython [11]是基于Python的工具使用的常见软件包。 CHOPCHOP和WU-CRISPR分别使用SVMlight [12]和LibSVM [13]。两者都是支持向量机(SVM)的C实现。同样,sgRNAScorer2和TUSCAN利用SciKit-learn软件包中的机器学习模块[14]。 sgRNAScorer2的作者提供了293T细胞系以及hg19和mm10基因组的模型。
CHOPCHOP,GT-Scan,CRISPR-ERA,CCTop和CasFinder使用Bowtie [15]进行脱靶;类似于mm10db和带有Bowtie2的CT-Finder [16];而CRISPOR和CRISPR-DO利用Burrow-Wheelers算法(BWA)[17]。 GuideScan不依赖于外部工具来偏离目标,而是实现了特里(trie)结构来设计具有更高特异性的指南[18]。 FlashFry受益于其引导基因组聚合方法,该方法能够在数据库的一次传递中识别出脱靶位点。与BWA相比,FlashFry的性能更高,因为不匹配的数量和候选向导的数量增加了[19]。有趣的是,[20]发现Bowtie2缺乏快速识别具有两个以上不匹配的所有脱靶位点的能力,而Cas-OFFinder [21]是更合适的解决方案,但是更耗时。
CHOPCHOP,Cas-Designer,mm10db,CCTop和CRISPR-DO提供了指定注释文件的方法。对于每种工具,我们在每个测试中都提供了适当的注释。 CHOPCHOP利用注释来指示候选向导靶向的基因或外显子,并允许用户将搜索区域限制为特定基因。 Cas-Designer利用自定义格式的注释文件,该文件描述了给定基因组上每个外显子的开始和结束位置。这用于设计专门针对外显子区域的候选指导。 mm10db需要UCSC RefGene格式的注释文件,以便生成包含所有外显子序列的文件。 CCTop利用注释来根据它们与最接近的外显子的距离评估指南,并将结果作为自定义轨道传递给UCSC基因组浏览器。所选工具均不限于特定的基因组。所有这些都已经开发出来,可以提供任何生物的基因组。这包括个人基因组,例如,如果将这些工具应用于个性化医学等领域。如表1所示,DNA或RNA凸起仅受两种工具支持。我们所提供的任何工具均不支持遗传变异。
工具之间共享一些生物学规则,例如:避免多胸腺嘧啶序列[22],拒绝GC含量不适当的指南[23],计算并可能考虑单核苷酸多态性(SNP)的位置以及考虑次级指南的结构。大多数工具会报告所有已确定的目标(有时会在特异性和/或效率上打出分数),并依靠用户来确定指南是否适合使用,而mm10db会主动过滤指南,仅报告“接受的”指南(但“被拒绝”的目标以及被拒绝的原因仍可在单独的文件中找到)。 WU-CRISPR和sgRNAScorer2不会通过程序样式的编程来实现任何这些规则,而是利用从实验数据训练而来的机器学习模型。此外,由于一些工具的时代和CRISPR相关研究的迅速发展,这些程序规则的细节各不相同。例如,早期研究表明,与PAM相邻的10-12个碱基对(种子区域)比指南的其余部分更重要[1,24],但最近的研究与此矛盾,并提出了1至5个碱基对PAM附近的配对更可能具有重要意义[25]。最近的研究表明,种子区域中的特定基序在存在时可使基因敲除频率降低多达10倍[26]。在评估工具时,这些可能是研究人员想要考虑的因素。
三、结果
3.1 Output
每个工具生成一组相似的候选指南,但是,相同指南的开始和结束位置通常最多相差4个位置。可以看到这是由于某些工具截断了PAM序列,或者使用了从零开始的定位。在标准化期间将省略号串联起来,以将其截断。根据UCSC数据集,所有指南均与基于1的定位保持一致,但是,请注意,Ensembl和Bowtie均采用基于0的系统,从而说明了其在某些工具中的用法。其中有17种工具使用逗号或制表符分隔的值(分别为CSV,TSV)格式产生输出,这使得它们的结果易于用于任何后处理。其中五个工具(GuideScan,CRISPR-ERA,mm10db,GT-Scan,CCTop和SSC)没有提供标题行来指示每一列的含义。 GT-Scan有所不同,因为它生成了一个SQLite数据库,其中包含所有候选指南的表。被终止的测试不会产生输出,因为在终止之前尚未达到写入文件例程。
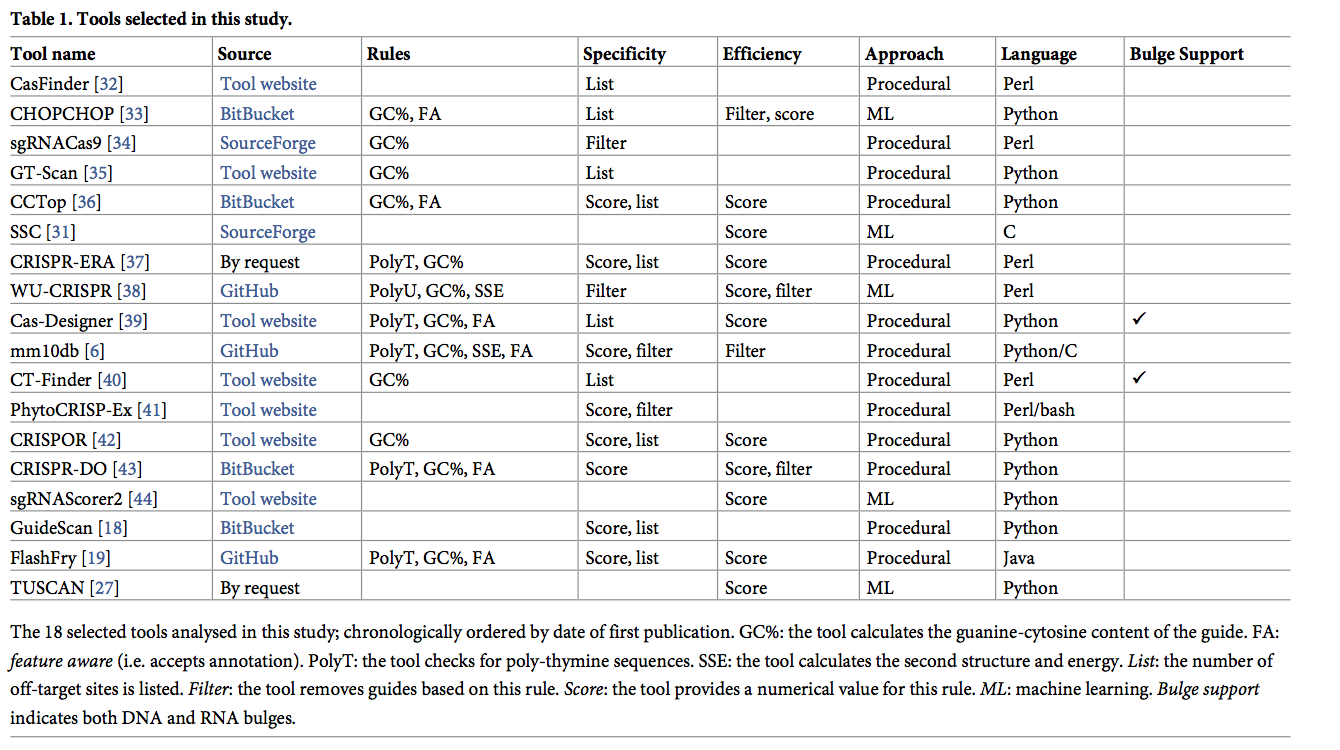
3.2 Output consensus
在这里,我们讨论了从chr19导出的数据集的工具之间的共识。我们专注于“ 500k”数据集,因为这是每个工具成功完成测试的唯一数据集。对于仍设法产生输出的工具,在较大的数据集上观察到了相似的结果。
我们将工具i和工具j之间的共识Ci,j定义为,i在j生成的指南的比例。 请注意,该值不是对称的:尽管两组参考线之间的交点没有变化,但Ci,j和Cj,i的分母分别是i和j生成的参考线数。
共识水平没有理想的绝对值。 如果有两种工具不能过滤目标,而只是报告所有候选站点,那么一个工具就可以达成完美的共识。 这没有说明目标的质量,但可以确认工具的行为符合预期。 另一方面,对于主动过滤目标的工具,可以期望共识水平较低,因为它们使用不同的标准来选择目标。 共识级别是分析的第一层,需要实验数据进行补充。
共识矩阵C如图1A所示,并着重指出大多数工具不会过滤目标。他们报告所有可能的目标,有时还会给出分数。这导致方法之间的高度共识,通常为95%或更高。另一方面,CHOPCHOP方法检查在第二十个核苷酸处是否存在鸟嘌呤。如果强制执行,它将仅报告其他工具产生的目标的约四分之一。 CRISPR-DO去除了含有多胸腺嘧啶序列的那些和具有不良脱靶作用的那些。 PhytoCRISP-Ex仅将满足以下两个规则的指南视为可能的靶标:(i)它们最多具有两个错靶位点,输入基因组中其他任何地方只有一个错配,以及(ii)其种子区(最后15个)碱基(包括PAM序列)是唯一的。然后检查这些潜在的指南中是否存在针对预先选择的常见限制性酶的限制性酶切位点。 WU-CRISPR与其他工具的共识最多为34.6%。这是由于该工具在使用SVM模型进行评估之前应用了预过滤器来消除指南。一些预过滤器包括:折叠和装订属性以及特定位置的指南内容。之后,该工具仅报告与模型匹配的指南。
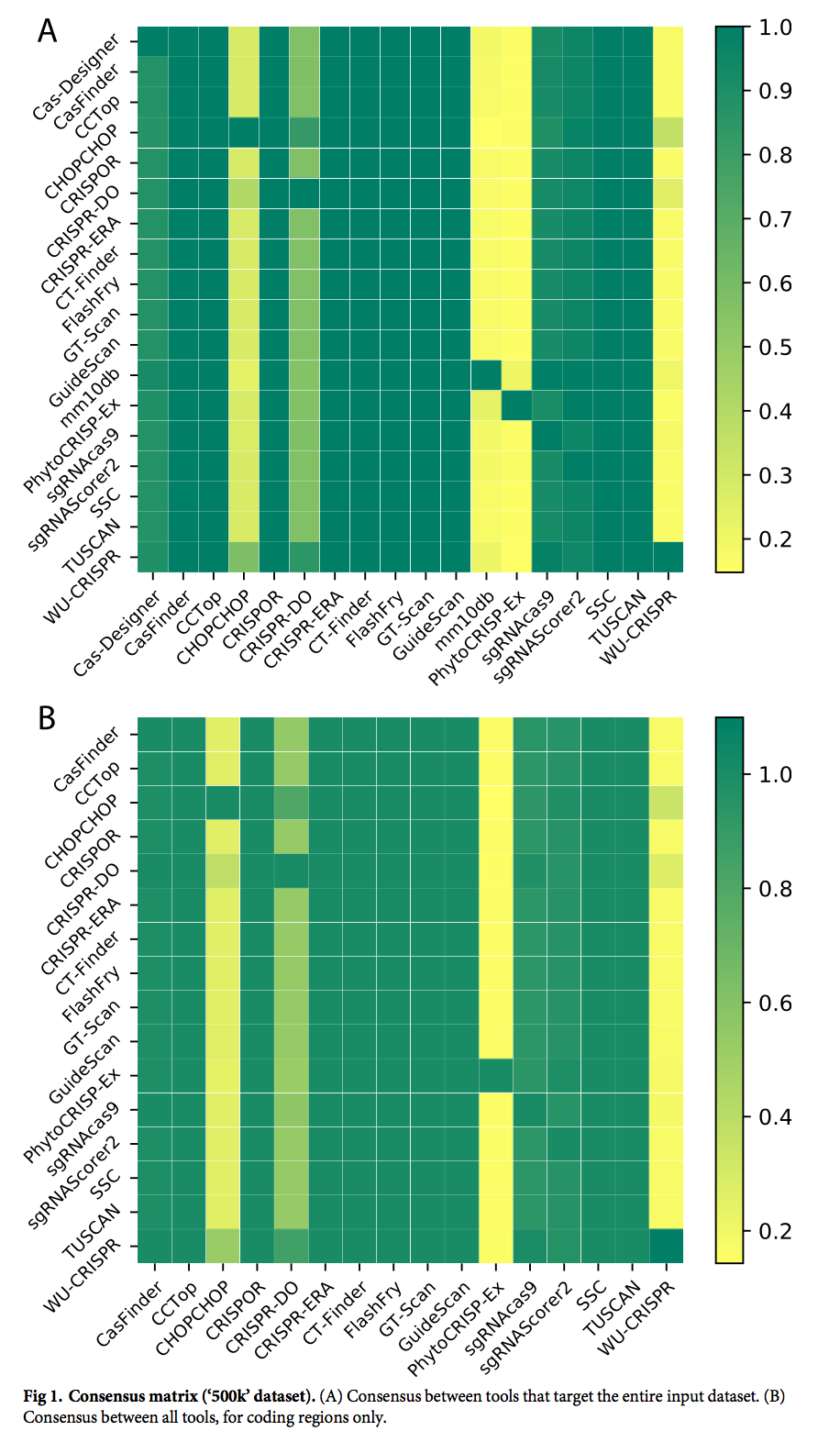
五个工具(CHOPCHOP,Cas-Designer,mm10db,CCTop和CRISPR-DO)聚焦,或 提供重点关注识别外显子上目标的选项。图1B显示了所有工具中这些区域的共识矩阵。和以前一样,在不拒绝目标的工具之间存在着很高的共识。对于更严格选择要报告目标的工具(CHOP-CHOP和mm10db),共识仍在15%至30%之间。有趣的是,CHOPCHOP和mm10db之间的共识很低。这突出表明工具正在使用不同的选择标准,从而导致识别不同的目标。
只有一个工具(mm10db)提供了详细的报告,以确定拒绝的原因。这些原因可用于分析其他工具的输出,并获得有关其行为的一些见解。值得注意的是,mm10db会顺序应用过滤器。例如,如果基因组中包含多个完全匹配的指南,则可能由于其GC含量而被拒绝。结果仍然是有益的。主要原因是较高的GC含量(约62%),较差的二级结构或能量(19%)以及多次完全匹配(16%)。脱靶得分是最后一步,因此尽管严格,但它必然会占拒绝的一小部分。在此,它甚至变得更小(<0.05%),因为小的输入大小限制了具有大量相似脱靶位的风险。
从图1中可以知道,mm10db拒绝了其他工具提出的指南中的83%。 对于每种工具,我们还探讨了mm10db给出的拒绝原因。 图2总结了“ 500k”数据集。 GC含量引起的排斥是最重要的原因,其次是二级结构或能量。

重要的是,由于基因组中的多个精确匹配,mm10db拒绝了这些工具提出的许多指南(平均约10%)。例如,在分析“ 500k”数据集时,由10个工具报告了指南CTCCTGAGTGCTGGGATTAAAGG,该数据集包含五个基因座处的指南作为完美匹配。本指南仅针对chr19上的两个不同基因:chr19:10,907,131-10,907,153的Prpf19和chr19:10,201,455-10,201,477的Gm10143。理想情况下,由于缺乏特异性,该指南不会出现在工具的输出中:没有实际的应用程序可以使用该指南。此外,没有任何一种工具能够通过报告k个不匹配的位点数量(k≤5)来处理特异性,实际上并没有为该指南报告正确的脱靶位数量。 Cas-Designer和GT-Scan报告完美匹配为零。 CHOPCHOP报告了50个完美匹配(CHOPCHOP使用Bowtie来完成此任务,但是仅限于报告50个对位)。使用Bowtie(-v 0 -a)将本指南与完整的chr19对齐,我们找到了241个完美匹配。 PhytoCRISP-Ex没有报告任何我们后来鉴定为在基因组中具有多个匹配项的指南。这是因为,作为mm10db,严格筛选出可能具有偏离目标的部分匹配的指南。
图2还显示,对于报告所有指南的工具,mm10db并未考虑(即既不接受也不拒绝)其输出的大约四分之一。 这是由于正则表达式用于提取候选项([ACG][ACGT]GG),它排除了以T开头的候选项,而其他工具则不这样做。
3.3 实验数据集
然后,我们考虑了过滤目标或提供可预测效率的得分的工具的性能。我们创建了两个人工基因组,每个基因组包含指导序列,可提供有关其效率的实验数据(请参见数据准备部分)。我们基于这些原始基因组的原始数据集:Doench和Xu。请注意,此处我们仅关注评估效率是否正确预测。因此,使用人工输入至关重要。如果我们针对整个基因组对指南进行评分,则可能由于与预测效率无关的其他因素(例如脱靶因素)而被拒绝。由于偏离目标的风险而被拒绝的指南,将告诉我们有关工具预测该指南是否有效的能力的信息。通过使用人工基因组,我们可以确保每个指南绝对没有偏离目标的风险,因此可以专注于效率预测。在这种情况下,一个完美的工具将接受实验证明有效的所有指南,而拒绝所有效率不高的指南。
结果如图3和图4所示。在我们的分析中,我们考虑了工具的精度,这是根据实际经验被认为是有效的,被预测为有效的指南所占的比例。
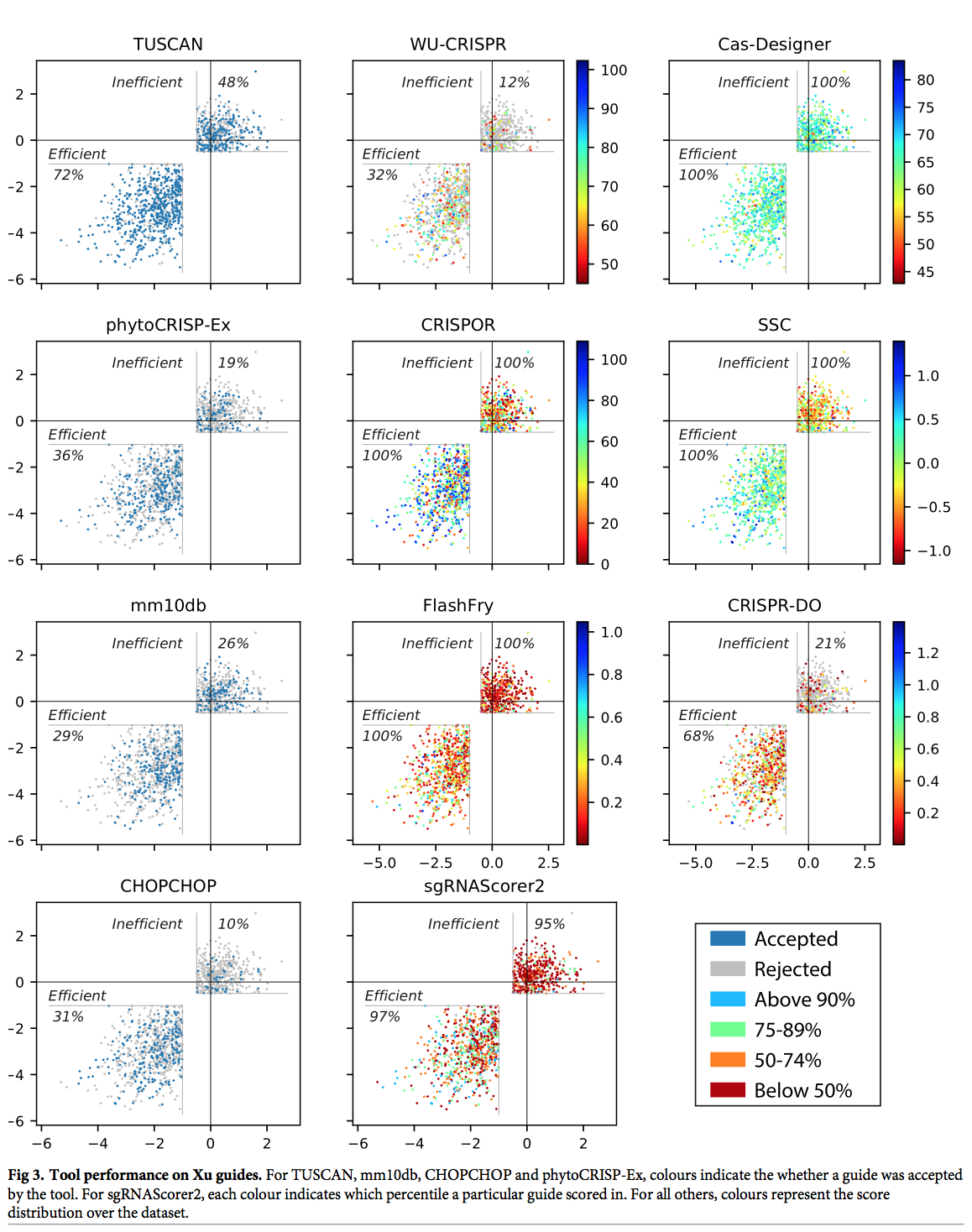
对于Xu数据集,CHOPCHOP接受了273条指南,其中84.3%是“有效的”,而mm10db接受了330条指南,其中65.2%是“有效的”。如果将CHOPCHOP的在20位上具有G的规则纳入mm10db,则其“有效”指南的比例将达到84.4%。结果还凸显了考虑多种选择方法的价值:mm10db识别的“有效”指南中,只有25.1%被选择为CHOPCHOP。
sgRNAScorer2用户手册指出分数应相对使用,而不是绝对分数:分数越高,预测的活动越好,但是没有阈值可以对预测的效率和低效率进行分类。 因此,图3中使用的第50,第75和第90个百分位仅用于可视化。 取而代之的是,我们提取了每对(a,b)指南,其中在实验上将a视为“有效”,将b视为“无效”,然后检查a的预测活动是否高于b。 其中76.8%的情况如此。
CRISPR-DO和WU-CRISPR没有报告人工基因组中提供的所有指南。 对于具有得分的指南,工具的精度分别为87.3%和81.8%。
对于所考虑的11种工具,总体共识非常低。 所有工具都只选择了整个数据集中的一个指南。
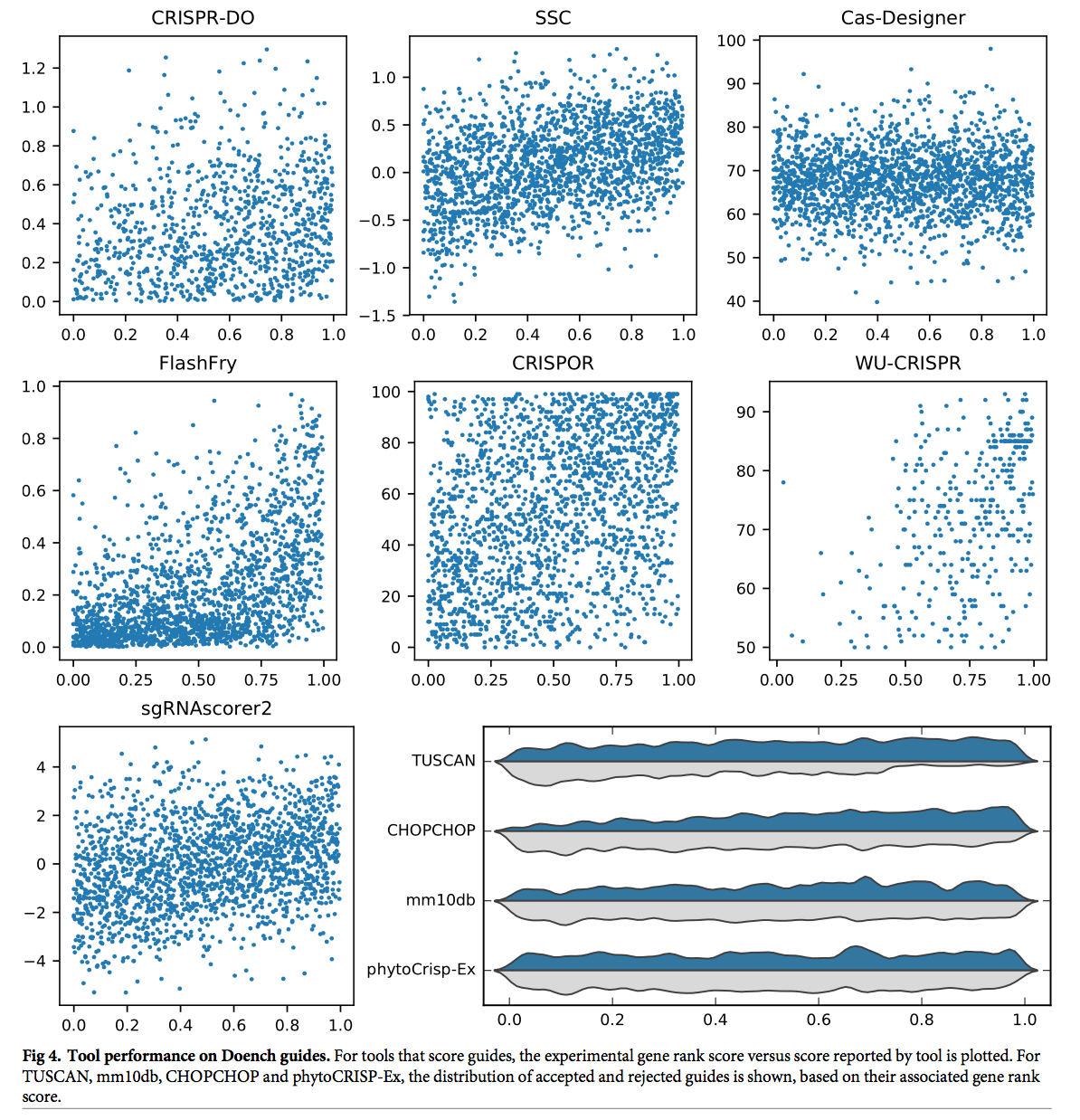
对于Doench数据集,提供了来自9个成绩单的指南,并且仅将每个成绩单前20%的指南视为有效[28]。 对于此数据集,我们发现与Xu相比,每种工具的精度都明显较低。 这是可以预期的,因为在本研究中使用高阈值来定义效率。 只有两个工具的precision高于50%:在该数据集上经过训练的WU-CRISPR,也使用最初从该数据集派生的评分方法的FlashFry。 TUSCAN在此数据集上进行了部分培训,但精度为24.5%。
如表2所示,所有其他工具的precision在20.2%(Cas-Designer)和30.4%(CRISPR-DO)之间。只有两个工具(mm10db和CHOPCHOP)的accuracy在65%以上。
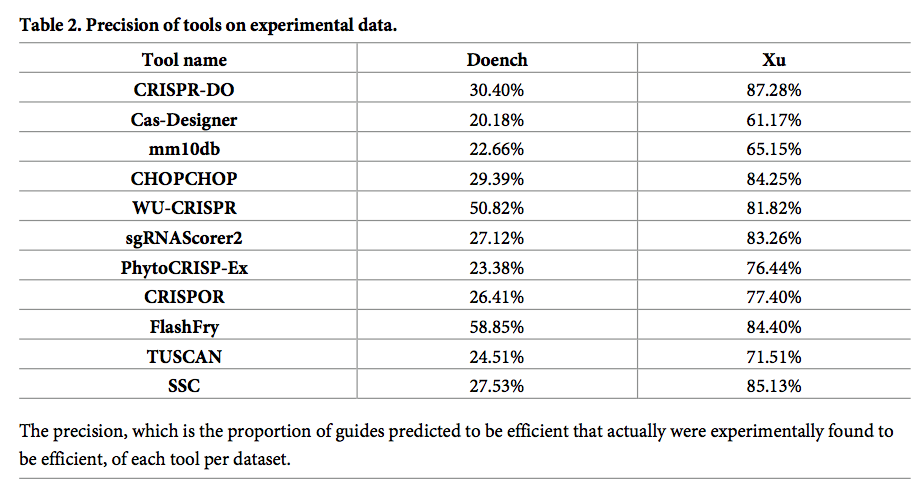
图4显示了该数据集上每种工具的性能。 对于提供分数的工具,我们将其预测作为基因等级百分比的函数。 对于FlashFry,具有较低基因排名百分比的指南通常得分较低。 WU-CRISPR在许多具有较高基因等级百分比的指南中均获得了高分。 TUSCAN接受大多数有效的指南。 对于SSC和sgRNAScorer2,随着基因等级的增加,较高分数的趋势非常微弱。 方位角评分(由CRISPOR报告),CRISPR-DO和Cas-Designer没有显示任何明显的趋势。 对于接受或拒绝指南的工具,我们显示了这两个类别在基因等级百分比上的分布。 对于mm10db,排名较高的指南更有可能被接受,而排名较低的指南则更有可能被拒绝。 对于CHOPCHOP,这不是很明显,对于phytoCRISP-Ex没有趋势。
再次,总体共识很低; 十一个工具在整个数据集中仅选择了三个指南。
3.4 计算性能分析
尽管最初介绍方法时常常被忽略,但是它们的计算性能仍然是重要的考虑因素。 CRISPR在许多应用中可能至关重要:大量输入基因组,获得结果的时间限制,大量非参考基因组进行分析等。
就时间要求而言,我们的假设是,任何有效的评分或过滤都将与要处理的靶标数量成线性比例(因为它们是单独评估的),因此与输入基因组大小几乎呈线性比例。但是,对于特异性分析,每本指南都需要与其他候选文献进行评估,这可能导致二次增长。另一个可能的限制是工具的内存需求。
我们的结果显示在表3和图5中。只有五个工具成功完成了四个测试:CasFinder,CRISPR-ERA,mm10db,GuideScan和CRISPR-DO。 GuideScan仅报告所有可能的目标,没有对预测效率进行评分,因此可以很快。 CasFinder和CRISPR-ERA提供得分,但在整个数据集中均相同,因此无法提供信息。另一方面,mm10db通过许多过滤器运行候选指南,并为脱靶风险提供有意义的评分,因此其速度值得注意。
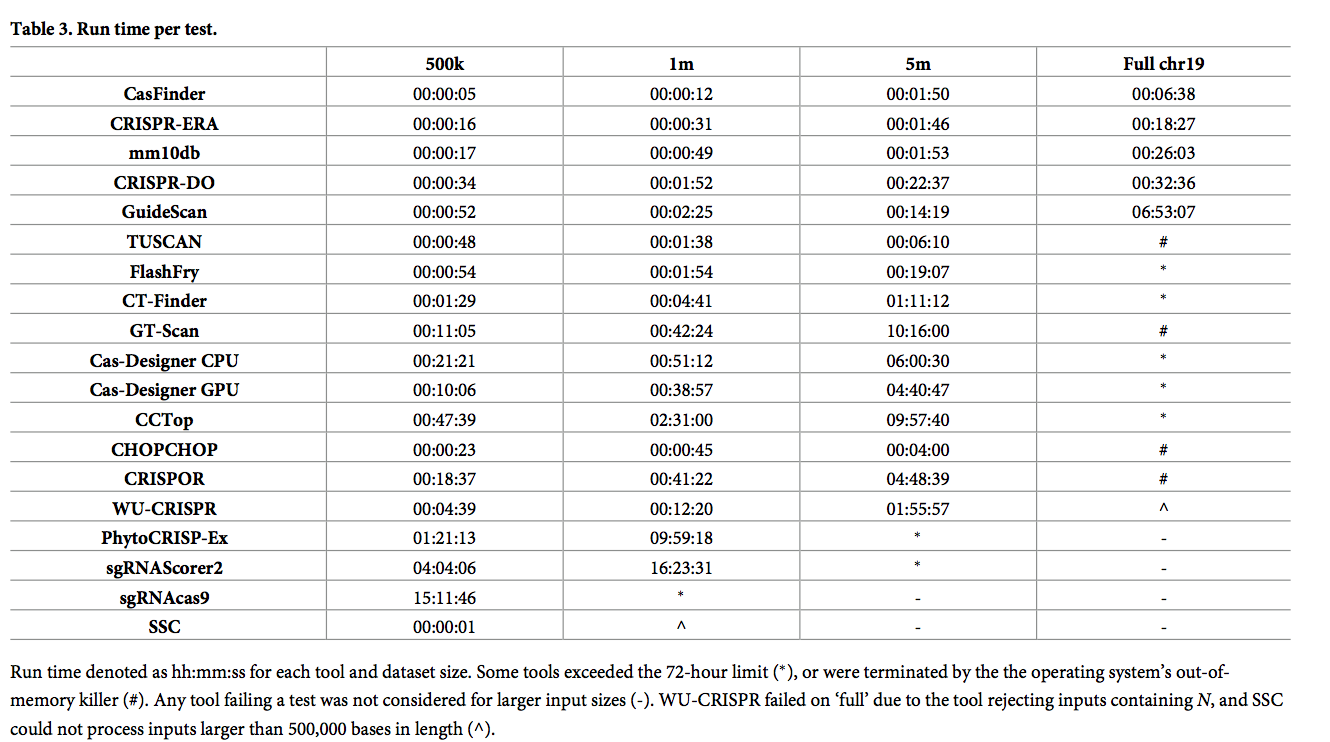
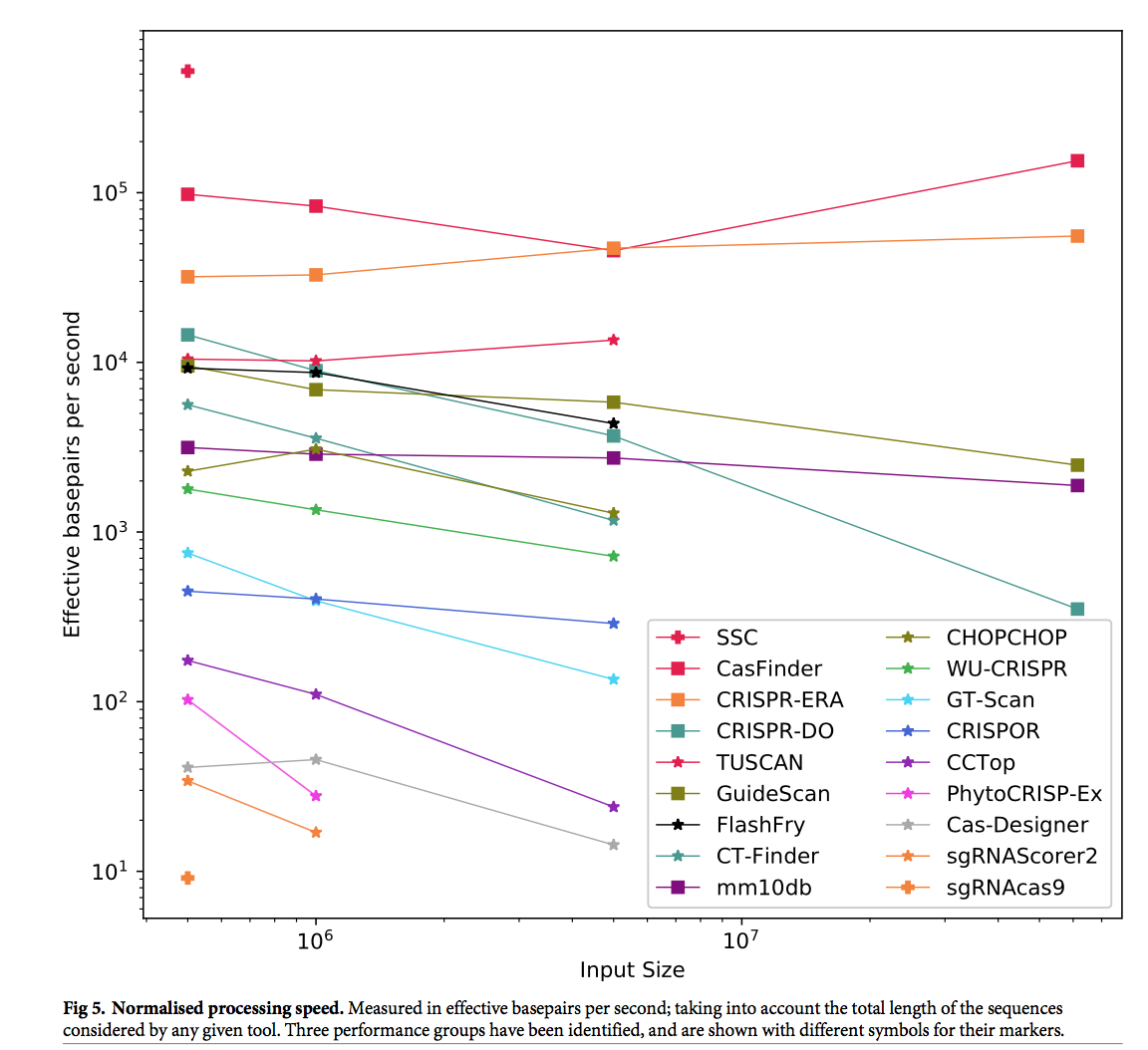
CHOPCHOP饱和了物理内存和分配的交换空间(由SBS监控),导致内存不足的杀手(OOMK)终止了其在全部19个染色体(约占小鼠基因组的2%)上的运行。 sgRNAScorer2的运行速度非常慢,需要花费4多个小时来处理500k数据集(而其他工具则需要5到21分钟之间的时间)。在不到3天的时间内,它无法处理5m数据集。 FlashFry在“满”以下的测试中表现不错,但是,在完成最终测试时,Java虚拟机已耗尽内存。
其他四个工具都不能满足时间限制,它们全部位于第19号染色体上(Cas- Designer,CT-Finder,GT-Scan和CCTop)。 CRISPOR,GT-Scan和TUSCAN的内存问题类似于CHOPCHOP的内存问题,并在同一数据集上终止。
作为分析的一部分,我们还考虑了多线程的使用。 CHOPCHOP,GuideScan,mm10db,FlashFry和sgRNAScorer2是仅有的实现多线程例程的工具。 CHOPCHOP和GuideScan不允许用户指定要产生的线程数,而是根据CPU内核数来产生线程。 mm10db提供命令标志以指定其自身和Bowtie的线程数;我们分别指定了128和8个线程。 Cas-Finder,CT-Finder和CHOPCHOP在单线程模式下使用Bowtie。
我们在GPU模式下运行Cas-Designer,发现这可以提高性能。 但是,这还不足以允许在时限内处理整个数据集。理想情况下,未来的工具(或此处基准的工具的未来版本)也应利用GPU资源。
SBS监视物理内存并分配交换空间使用情况。 CRISPOR,CHOP-CHOP,GT-Scan和TUSCAN都饱和了这两个内存空间,导致内存不足杀手(OOMK)终止了整个数据集测试中的每个工具。 Cas-Designer满足了交换空间的要求,但是在同一测试中并未触发OOMK。
四、讨论
已经提出了大量工具来帮助设计CRISPR-Cas9指南。 其中许多,特别是我们研究中的三个,已经在实验上成功使用:CasFinder已用于设计人类基因组以靶向特定基因的指南[29],CRISPR-ERA已用于设计靶向HIV-的指南 1个病毒基因组[30]和mm10db已用于小鼠基因组的全基因组分析[6]。
但是,它们的性能几乎没有比较。 本文通过对18种工具的计算行为及其产生的基准进行基准测试来解决这一差距。 我们的结果表明,只有五个工具(CasFinder,CRISPR-ERA,CRISPR-DO,GuideScan和mm10db)可以声称能够快速分析大量输入,并且可以轻松地处理整个基因组,尤其是较大的基因组。
许多工具在设计指南时不是选择性的:它们会报告所有候选者,因此在它们之间具有很高的共识。另一方面,如表2所示,有11种工具提供了指南效率的清晰预测。我们在经过实验验证的指南的两个集合中评估了它们的性能。所有11种工具都为Xu数据集提供了大多数有效的指南,但为Doench数据集却提供了很少的有效指南。对于这两个数据集,工具之间的重叠很少。这表明这些工具确实具有检测某些有效指南的能力,但是它们并不完全精确,并且召回率仍然很低。需要改进的方法。
两者合计,结果表明
- mm10db目前在速度和全基因组分析指南的过滤/评分之间达到了最佳平衡。
- 当研究少数基因时,慢速但更具预测性的CRISPR-DO可能是首选。
- CHOPCHOP在缩放到整个数据集时遇到问题,但否则也将是一个不错的选择。
重要的是,结果还强调了需要进一步完善CRISPR-Cas9指南设计工具。该基准为优化计算性能和组合多种设计方法的未来工作提供了明确的方向。
五、材料和方法
考虑到规则和实施的范围,对基准设计工具进行基准测试并比较其性能至关重要。 在本节中,我们描述如何根据计算需求,功能和输出评估每种工具。
5.1 资料准备
我们基准测试的初始数据基于GRCm38 / mm10小鼠基因组组装,可通过加利福尼亚大学圣克鲁斯分校(UCSC)获得。我们下载了chr19,并提取了三个长度递增的数据集:500k,1m和5m核苷酸,所有数据都从位置10,000,000开始。这些数据集以及整个染色体都用于测试。
对于这四种配置中的每一种,我们创建了任何工具所需的所有文件:自定义批注文件(来自可通过UCSC获得的refGene表),2位压缩文件,Bowtie和Bowtie2索引以及Burrows-Wheeler Aligner文件。
为了补充这些数据集,我们还使用了两个指南集,这些指南可提供实验数据[28,31]。徐(Xu)收集了1169份用于筛选实验的指南,根据对基因敲除的分析,其中731份被认为是“有效的”。另一个Doench包含371个“有效”指南和1470个“无效”指南。了解指南的实验质量可以进一步了解评估指南的计算技术的质量。
我们构建了两个人工序列,其中包含来自每个数据集的引导,相隔50 N,以确保无法检测到意外的重叠目标。和以前一样,我们生成了此输入所需的所有支持文件。
这些工具并未针对特定生物进行优化,并且选择mm10作为初始测试,还是选择人类细胞系的数据作为实验验证,都不会影响结果。
我们的数据集可在S1数据集中找到。
5.2 绩效基准 Performance benchmarking
我们的软件基准脚本(Software Benchmarking Script ,SBS)工具在Python 2.7中实现,并使用“过程和系统利用率”(Pro- cesses and System Utilisation, PSUtil,版本> = 5.4.4)模块来进行特定于过程的系统资源监视。启动时,要求用户通过命令行标志传递bash命令和输出目录。审计例程在执行bash命令之后开始。在每个轮询事件(PE)上监视父进程和所有后代。
记录当前的挂墙时间,CPU和内存使用情况,磁盘交互(DIO),线程数和子代数。壁挂时间(Wall-time)比CPU时间更可取,因为它是人类可以感知的完成时间。 SBS报告瞬时驻留集大小(RSS)使用情况和虚拟内存使用情况。 DIO包括读/写操作的数量和大小。在每个PE处,将计算父数据和子数据的汇总并将其写入文件。此外,还会记录启动每个子进程的bash命令。
PE例程继续进行,直到父进程结束或超过72小时为止。施加此限制是因为我们旨在讨论有潜力进行全基因组分析的工具,而在此限制内无法分析chr19(占mm10总大小的2.25%)的工具被认为不适合该任务。
所有测试均在具有Intel Core i7-5960X(3.0 GHz),32 GB RAM,32 GB分配的交换空间和Samsung PM87 SSD的Linux工作站上执行。我们使用了Python v2.7和Perl v5.22.1。该机器超出了某些工作站的规格,但是,期望用户需要一台类似或更高的机器才能进行全基因组分析。
SBS可以在GitHub上找到,网址为https://github.com/jakeb1996/SBS。
5.3 输出归一化和比较
每个工具都有自己的输出格式,因此我们将结果归一化为:工具名称,候选指导序列,起始位置,终止位置和链(以CSV格式写入文件)。 在此过程中,根据UCSC数据集,将起始值和结束值与基于一的位置对齐。 将省略号连接到缺少PAM序列的指南。
为了确定哪些工具共享公共指南,脚本将汇总所有非重复的指南,并记录哪个工具产生了随后出现的指南。 当3’位置相等且据报道它们靶向同一条DNA链时,该指南被视为与先前观察到的指南重复。 一个单独的脚本分析了每个标准化的指南,以确定它是否针对基因编码区(基于UCSC注释数据)。
参考资料
- Citation: Bradford J, Perrin D (2019) A benchmark of computational CRISPR-Cas9 guide design methods. PLoS Comput Biol 15(8): e1007274. https://doi.org/10.1371/journal.pcbi.1007274 获取文献
