【6.5.2】优化的sgRNA设计可最大化crisPr-cas9的活性并最小化脱靶效应
基于CRISPR-Cas9的基因筛选是生物学中强大的新工具。 通过简单地改变单向导RNA(sgRNA)的序列,人们可以相对容易地将Cas9重新编程为靶向基因组中的不同位点,但是单个sgRNA的靶向活性和脱靶作用可能差异很大。 在这里,我们使用最近设计的sgRNA设计规则来创建人类和小鼠全基因组文库,进行正负选择筛选,并观察到这些规则的使用产生了改进的结果。 此外,我们分析了数千个sgRNA的脱靶活性,并开发了一种指标来预测脱靶位点。 我们从大规模的经验数据中整合了这些发现,以改善我们的计算设计规则,并创建优化的sgRNA库,以最大程度地提高靶标活性并最小化脱靶效应,从而实现更有效的遗传筛选和基因组工程。
一、前言
Cas9蛋白是一种RNA定向的DNA核酸内切酶,是操纵基因组的强大工具。易于编程的Cas9(CRISPR相关蛋白9)使基于CRISPR(聚簇的,规则间隔的,短回文重复序列,clustered, regularly interspaced, short palindromic repeats)的遗传筛选成为可能,从而鉴定出完善的基因并提供了对多种表型的基因功能的新颖见解。初始文库的设计几乎不了解sgRNA活性规则,这是一个关键的设计参数,因为解释筛选数据需要靶向同一基因的多个sgRNA之间具有一致性,以区分真假和假阳性。非活性和非特异性sgRNA会降低文库的有效基因覆盖率和命中列表的准确性。
许多研究表明,Cas9脱靶活性取决于sgRNA序列和实验条件。这些研究提供了对特异性决定因素的定性但不完整的理解。寻找可通用的模式非常具有挑战性,需要大量数据集来对大量可能的不完美的sgRNA-DNA相互作用进行充分采样,以揭示预测脱靶活性的序列特征。在这里,我们根据我们先前发布的预测目标效率的规则,介绍了人类和小鼠全基因组sgRNA文库的设计和表征。基于新库生成的筛选数据和脱靶活性的大规模评估,我们开发了用于脱靶和脱靶活性预测的改进算法,从而可以进一步优化我们的全基因组文库。
二、结果
2.1 使用Avana和Asiago库进行遗传筛选
以前,我们检查了1,841个sgRNA的活性,以确定导致功效增强的序列特征,并制定了改进sgRNA设计的规则(规则集1)【14】。我们在分别名为Avana和Asiago的人类和小鼠全基因组库中实施了这些规则,并在表型筛选中测试了它们的性能。我们根据三个标准:
- 规则集1得分,
- 蛋白质编码区域内的特异性
- 基因内的目标位点位置(补充表1-3)
每个基因选择了六个sgRNA。先前发布的GeCKO6,和Koike-Yusa等人,库的规则集1分数的分布类似于零分布,因为这些库不是根据目标标准设计的(图1a)。 Wang et al库纳入了提高sgRNA活性的早期规则,并且规则集1得分更高。根据设计,Avana和Asiago库是最丰富的预计具有活性的sgRNA。
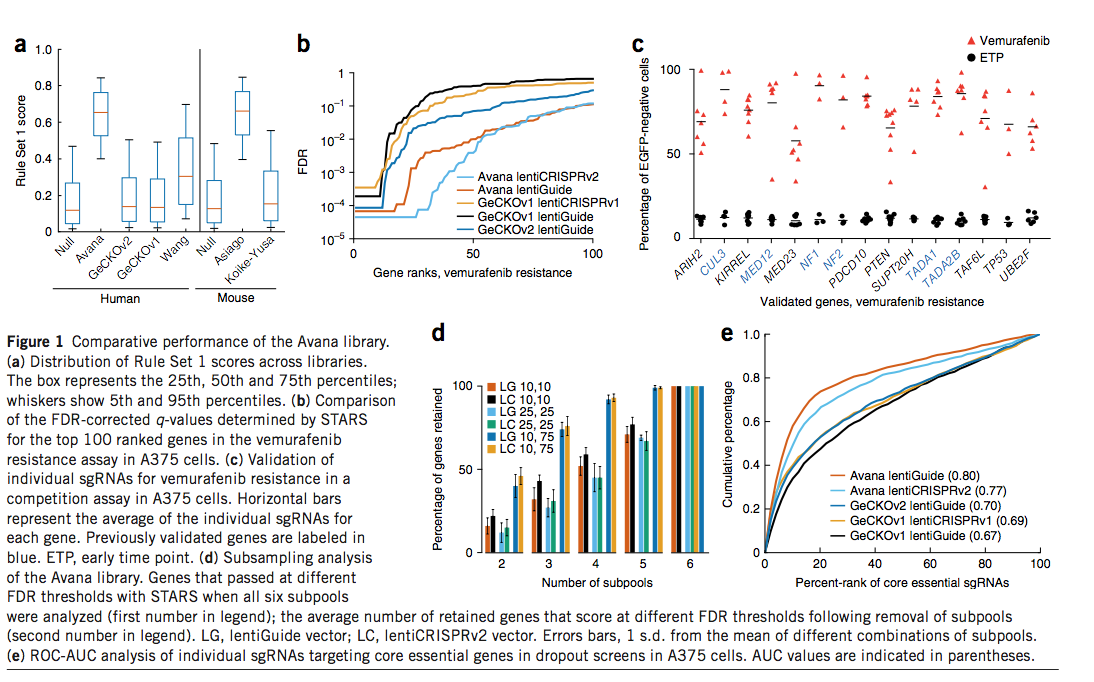
我们首先使用一个完善的筛选系统对我们的文库进行了测试,该系统对A375黑色素瘤细胞具有维拉非尼(Zelboraf)的耐药性,该细胞携带BRAF V600E突变并且对MAPK途径抑制敏感(补充图1和2)6,16,17 。 我们以低感染度进行生物复制,转导了六个Avana亚库中的每一个,每个亚库每个基因包含1个sgRNA,并用GeCKOv1和GeCKOv2文库进行了相同的筛选。 对于用Avana文库感染的细胞,我们还应用了MEK抑制剂selumetinib18,19,这是先前在此抗性模型中检查过的另一种小分子(补充图3)。 我们通过相对于它们在质粒DNA池中的丰度的log2倍数变化对sgRNA进行排名,并根据两次重复的结果得出平均排名(补充表4)。
我们首先使用RIGER算法来分析GeCKOv1,GeCKOv2和Avana筛选(补充表5和补充图4)。但是,带有RIGER的加权和选项仅包含来自针对基因的前两个扰动的信息,而没有其他sgRNA提供的有关命中列表特异性的其他证据。因此,我们开发了STARS,这是一种替代的基因排名系统,可产生假发现率(FDR),奖励那些在检测中有很高sgRNA活跃的基因,类似于MAGeCK算法。使用STARS,我们观察到先前验证可赋予维罗非尼耐药性的6个基因(CUL3,MED12,NF1,NF2,TADA1,TADA2B)均在FDR <1%时得分(补充表6)。
我们直接比较了lentiGuide向量中这三个库的性能。在FDR <10%的情况下,提名了27个基因被GeCKOv1命中,而60个基因被GeCKOv2评分(图1b),这可能是由于文库大小而增加的,因为GeCKOv1平均每个基因3至4个sgRNA,而GeCKOv2每个基因包含6个sgRNA。 。 Avana对FDR <10%的评分为92个基因,每个基因还包含6个sgRNA。同样,在慢病毒CRISPRv2载体中筛选了用Avana文库评分的94个基因,在这两个载体中FDR <10%的36个基因评分。
我们检查了新的维罗非尼耐(vemurafenib)药性调节剂的数据。在Avana文库中,抑癌基因PTEN,TP53和RB1的FDR <1%,而在GeCKOv2文库中,这些基因的FDR分别为78%,7%和2%(补充表6)。因此,我们注释了PanCancer基因的命中列表,PanCancer基因是经过高度精心策划和验证的集合,其中包含许多基因,这些基因的丢失可能会恢复MAPK途径或激活其他生存途径(补充表7)。我们观察到,与任一版本的GeCKO相比,Avana库识别出的PanCancer基因更多。在FDR <10%的情况下,我们鉴定出四个具有GeCKOv1的PanCancer基因(P = 1.1×10−5,超几何分布),六个具有GeCKOv2的基因(P = 2.2×10−7)和十个具有Avana的基因(P = 2.9×10− 11)。在selumetinib筛选中,Avana鉴定了20个基因(P = 4.6×10-24),包括在可比的vemurafenib数据集中以相同阈值鉴定的10个基因中的9个(补充表8和9)。
我们使用竞争分析法验证了一些新发现的匹配项。将携带单个sgRNA的细胞与EGFP标记的细胞混合;如果该sgRNA提供了选择性的生长优势,则EGFP阴性细胞会随着时间的流逝而富集,如通过流式细胞仪测量的。我们用九种阳性对照sgRNAs(靶向NF1,NF2,CUL3)测试了该检测方法;十个没有已知靶标的阴性对照sgRNA;以及五个sgRNA的新产品TP53。当不使用vemurafenib进行培养时,含sgRNA的细胞比例会略有下降,在所有sgRNA上平均下降8.4%(补充图5)。在存在vemurafenib的情况下,带有阴性对照sgRNA的人群死亡,无法通过流式细胞仪评估,而阳性对照和TP53 sgRNA分别平均占总人口的93%和82%(补充)。图5)。在对六个先前验证的基因和九个新近鉴定的基因进行的相似竞争分析中,加入维罗非尼后含sgRNA的细胞的平均代表范围分别为80%至90%和58%至84%,从而验证了可重复性Avana主屏幕(图1c)识别的所有九个选定命中的结果。这些新验证的命中数个代表先前确定的复合物的其他成员。
尽管每个基因筛选更多的sgRNA可提高文库检测命中的能力,但较大的文库需要进行大规模筛选,从而减少了可以筛选的条件数量,并且限制了细胞数量有限的模型。我们进行了二次抽样分析,以确定减少sgRNA的数量如何影响vemurafenib筛选的命中列表。在lentiGuide载体中筛选的Avana库鉴定出FDR <10%时有92个结果。对于四个每个基因一个单sgRNA子池的组合,在相同的FDR下平均可回收52%的基因(图1d)。在宽松的FDR阈值<75%时,用4种sgRNA回收了92%的基因。对于在慢病毒CRISPRv2中筛选的Avana文库(93%)和在慢病毒指南(93%)和慢病毒CRISPRv2中的selumetinib筛选(93%),观察到的比率相似(补充图6)。该观察结果提出了一种有用的筛选策略,尤其是对于阵列筛选或合并模型,因为后者的规模扩大成本高昂或令人望而却步:以基因组规模对少量sgRNA进行初次筛选,使用相对宽松的分界值来进行命中选择,然后对每个基因使用额外的sgRNA对数百个主筛选命中进行次筛选。
接下来,我们针对阴性选择,靶向增殖必不可少的基因的sgRNA的消耗检查了文库性能。与阳性选择不同,在阳性选择中,小部分细胞可以大量富集,而dropout筛查则代表了更具挑战性的筛查方式,因为接受扰动的大部分细胞必须显示表型才能得分。我们在A375细胞中使用Avana,GeCKOv1和GeCKOv2文库(补充图7以及补充表10和11)进行了活力筛选,并分析了针对291个核心必需基因的精选集的sgRNA(补充表11)。为了能够比较不同大小的文库,我们对单个sgRNA进行了接收器操作特征-曲线下面积(ROC-AUC)分析。 Avana库的性能显着优于任一版本的GeCKO库,其AUC值分别为0.77和0.80,而GeCKO AUC值为0.67至0.70(图1e)。使用STARS,Avana在291个基因中鉴定了171个(占59%),而GeCKOv2在FDR <10%时鉴定了76个(29%)(补充图8和补充表12)。同样,使用基因集富集分析26,Avana库以比GeCKOv2更高的统计置信度确定了必需的细胞复合物和过程(补充图9)。结合来自Avana库和GeCKO库的数据,然后进行STARS分析,分别在A375细胞中的FDR <10%和<25%时确定了1,545和1,952个必需基因,该范围与最近的报告一致(补充表13)。
我们证实了在结肠癌系HT29细胞中Avana文库的负选择筛选性能得到了改善(补充表10和11以及补充图10)。在这里,我们筛选了Avana文库的前四个子池,并观察到核心必需基因的耗竭有了显着改善,尽管使用了较少的sgRNA:具有4个sgRNA的Avana鉴定了161个基因,而具有6个sgRNA的GeCKOv2鉴定了92个基因(补充表12)。
为了在不同的测定中进一步测试Avana库,我们在三种不同的细胞系中筛选了对嘌呤类似物6-硫鸟嘌呤的抗性。在A375细胞中,单个sgRNA占据了六个亚细胞池的每个,而所有六个靶向HPRT1都在该测定法中得到了很好的确立(图2a和补充表14)。每个亚库中的HPRT1 sgRNA的富集程度比排名第二的sgRNA至少高700倍,这表明该文库中的其他〜110,000 sgRNA均未导致HPRT1基因座处的显着脱靶活性。在HT29和293T细胞中,HPRT1 sgRNA高度富集,但靶向NUDT5也产生了6-硫鸟嘌呤抗性(图2a)。我们通过在这三种细胞类型中分别感染两种针对HPRT1和NUDT5的sgRNA的个体感染验证了这些观察结果(补充图11)。为了了解细胞系中NUDT5 sgRNA的差异富集,我们通过TIDE技术检查了基因组DNA。在没有选择的情况下,两种sgRNA均导致所有三个细胞系中NUDT5的修饰(图2b),这表明观察到的细胞系之间的表型差异并不是由于无效的基因编辑。
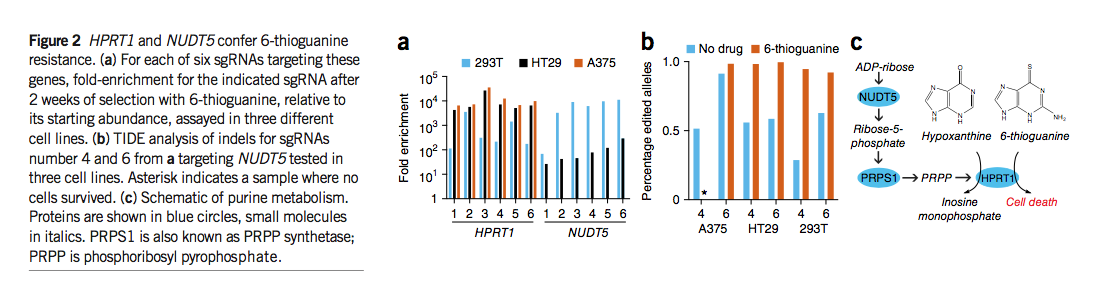
6-硫鸟嘌呤是嘌呤挽救途径中HPRT1的底物,替代次黄嘌呤并因此引起毒性(图2c)。 NUDT5在嘌呤合成中起着较早的作用,最终导致产生5-磷酸核糖基二磷酸酯(PRPP),这是HPRT1催化的肌苷一磷酸30的另一种底物。因此,NUDT5的消耗可能会阻止6-硫鸟嘌呤掺入的毒性作用(图2c)。 A375和HT29细胞之间表型严重性的差异可能与以下事实有关:A375细胞丢失了NUDT5基因的一个等位基因,并且可能上调了产生PRPP的替代手段。
最后,我们在干扰素信号传导模型中测试了Asiago小鼠文库,其中BV2细胞受到干扰素γ攻击,对存活细胞的STARS分析显示,丰富的成熟的干扰素信号传导介质(表1和补充表15)。十个最丰富的sgRNA中有九个以Jak1,Stat1,Ifngr1或Ifngr2为目标,而这些基因的所有四个sgRNA均在被筛选的79,641个sgRNA的前25个中得分。有趣的是,用人类同源物查询分子特征数据库,发现FDR <50%时鉴定出的线粒体基因显着富集(FDR = 8.99×10-12,超几何检验),以前没有干扰素信号传导。

2.2 sgRNA靶向活性的规则集2 (Rule Set 2 for sgRNA on-target activity)
我们最初使用1,841个sgRNA来模拟靶向活性,该sgRNA靶向流式细胞仪(FC数据集)检测到的三个人类和六个小鼠基因。为了改善预测,我们设计了一个平铺文库,该文库针对15个已知对vemurafenib,selumetinib,6-硫鸟嘌呤或依托泊苷具有抗药性的基因中的所有可能的NGG邻近原型间隔位(PAM)的位点。在A375细胞中,据报道有几个基因对依托泊苷(TOP2A,CDK6)和6硫鸟嘌呤(MLH1,MSH2,MSH6和PMS2)产生抗性命中,在其他细胞类型中,在条件此处测试的位置。对于产生抗性细胞的小分子,我们检查了sgRNA富集(图3a和补充表16)。如预期的那样,vemurafenib和selumetinib之间的单个sgRNA活性相关性很好(图3b)。这些靶向八个基因的2,549个sgRNA组成了RES(抗性)数据集。
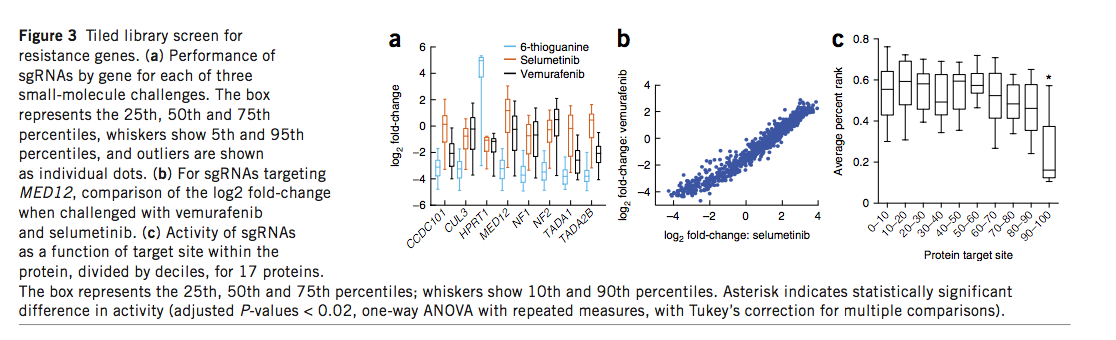
利用针对17个基因的4,000多个sgRNA的组合数据,我们检查了每种sgRNA的功效及其在蛋白质编码区域中的位置(补充图12)。初始设计指南建议针对N端附近。另外,Vakoc及其同事最近报道了当靶向几个特征充分的蛋白质的特定结构域时,功能丧失等位基因的产生得以改善。但是,对于这里考虑的17个基因,我们没有观察到蛋白质编码序列的离散区域,这些离散区域显然是基因失活的生产力更高的目标位点(补充图12)。总体而言,我们观察到只有10%的蛋白质编码区域的C端显示出活性的统计学显着降低(P <0.02)(图3c)。扩展的靶位点窗口可在sgRNA选择上提供更大的灵活性,既可优化靶上疗效,又可将脱靶潜力降至最低。
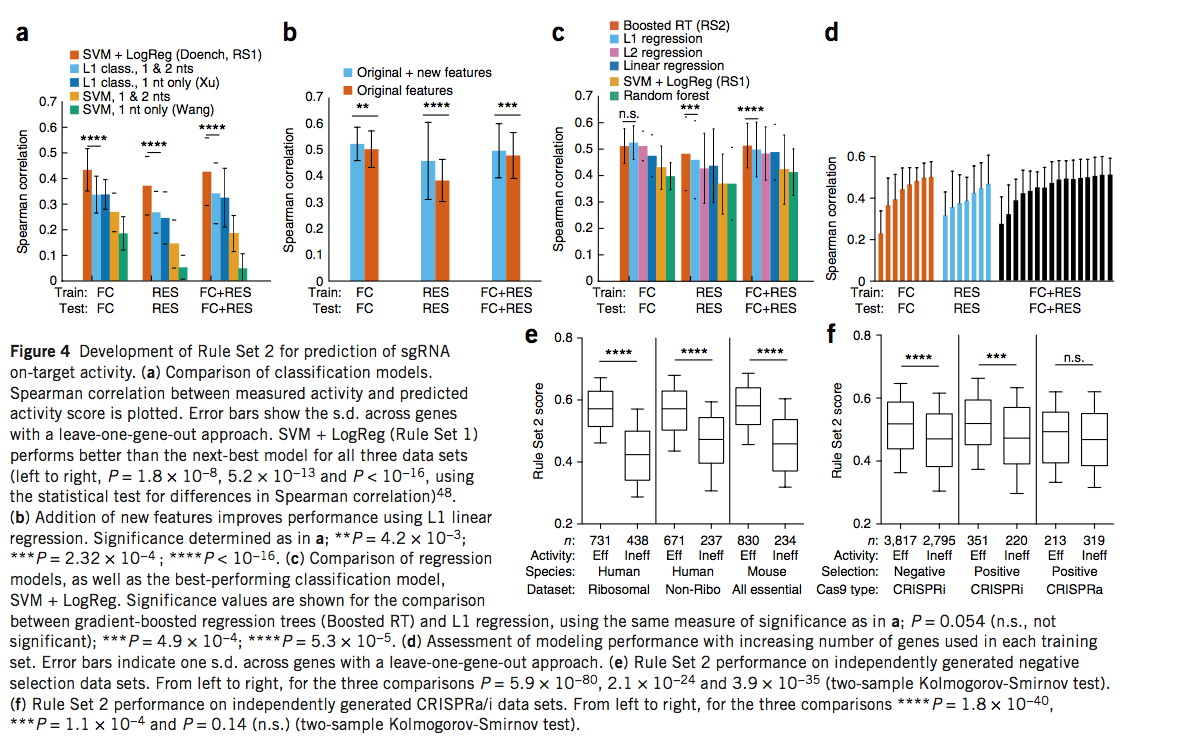
以前,要生成规则集1,我们将每个基因的前20%sgRNA视为高度活跃,并在20%与80%分类模型中进行训练以识别预测特征【14】。我们将使用支持向量机(SVM)进行逻辑回归的该模型的性能与应用于sgRNA活性预测,SVM和L1逻辑回归的其他基于分类的方法进行了比较,并使用了leave-one-gene-out方法。评估FC,RES和组合数据集后,我们观察到了SVM和逻辑回归的最佳性能(图4a和补充图13)。在这些数据集上,先前发现具有信息性的仅靠SVM或L1逻辑回归模型增强了二核苷酸特征,其性能比SVM加logistic回归差(图4a)。
将sgRNA活性离散化为20%和80%的类别(虽然方便)可能会导致大量信息丢失,因此我们使用归一化sgRNA等级探索了回归模型。我们最初保留了与规则集1,AUC,并观察到在具有相同特征的相同FC数据上训练时,L1正则化线性回归(连续回归模型)优于L1正则化logistic回归(离散分类模型);我们用Spearman相关性观察到了相似的结果,Spearman相关性是更适合回归模型的指标(补充图14)。这些结果建议进一步探索线性回归模型。
除了用于预测活动的建模方法外,另一个重要因素是feature set。以前,我们使用单核苷酸和二核苷酸位置特异性核苷酸,以及sgRNA的GC计数。我们假设其他特征,例如位置无关的核苷酸计数和sgRNA靶位点在基因中的位置,可以改善预测。生化和结构研究表明,sgRNA参与了与DNA的逐步(step-wise)关联,这表明局部热力学性质也可能有用。这些附加功能改进了所有数据集的L1回归模型(图4b)。提高sgRNA活性的微同源特性(Microhomology features,)可以单独预测,但添加到我们的最终模型中并不能提高性能。
由于回归模型和其他功能可以提供更好的结果,因此我们研究了五个不同回归模型的性能。在RES数据和组合数据集上,梯度增强的回归树模型表现出比所有其他模型更好的Spearman相关性,而对于FC数据集,其表现与L1和L2回归模型相当(图4c)。我们得出结论,在这里比较的那些当中,梯度增强回归树模型是最好的。在此模型中,sgRNA每个位置的二核苷酸和单核苷酸同一性继续对活动预测做出最大贡献,占Gini重要性的58%(衡量每个特征对整个模型贡献的重量)(补充图15和补充表17)。New features 做出了重要贡献:position-independent counts of single and dinucleotides,蛋白质内sgRNA的位置,以及不同区域的解链温度,分别占Gini重要性值为16%,13%和11%。为了衡量数据量是否使模型饱和,我们将基因系统地添加到训练集中,评估了性能,并观察了合并数据集的平稳性(图4d)。此外,当对所有成对的数据集组合进行训练和测试时,使用组合的数据集训练的模型在所有测试集上都产生了最佳结果(补充图16)。我们将梯度增强回归树模型与在组合数据集上训练的增强特征集称为“规则集2”。
为了进一步验证规则集2,我们评估了来自两个人类细胞系和小鼠胚胎干细胞的,独立的阴性选择数据集的性能,该数据集未用于构建或评估最终模型,这些数据由Xu及其同事策划。规则集1评分在所有三个数据集中均清楚区分了有效和无效的sgRNA,P值分别为1.4×10-32、1.8×10-16和1.1×10-11(两次样本Kolmogorov-Smirnov检验;补充图17)。在规则集2中,我们观察到了有效和无效sgRNA之间的更大区别,P值分别为5.9×10-80、2.1×10-24和3.9×10-35(图4e),说明了我们的建模方法的一般性。
接下来,我们检查了使用CRISPR激活(CRISPRa)和CRISPR干扰(CRISPRi)技术的屏幕中精选的数据集。在最大的CRISPRi数据集中,在阴性选择筛选中包含5,000多个sgRNA,规则集2在被分类为有效与无效的sgRNA得分分布上表现出最大差异(P值= 1.8×10-40,两个-样本Kolmogorov-Smirnov测试;图4f)。带有571个sgRNA的较小的正选择筛选的P值为1.1×10-4。在最小的数据集中,从阳性选择CRISPRa筛选中获得532个sgRNA,规则集2再次偏爱有效的sgRNA,但P值较高(0.14)。总之,这些观察结果表明CRISPR基因敲除与CRISPRa / i之间的共性,例如sgRNA与Cas9之间的相互作用以及sgRNA与DNA之间的相互作用,代表了规则集2预测能力的重要组成部分。
2.3 CFD评分可预测sgRNA脱靶效应
sgRNA的脱靶作用已在许多实验系统中进行了广泛研究,关于脱靶活性程度的结论也有很大差异【5】。 我们试图利用针对人CD33编码序列以及所有可能的sgRNA的文库,在哺乳动物细胞的典型合并筛选条件下了解脱靶效应,而与PAM无关。 对于具有规范NGG PAM的所有位点,除了完美匹配的sgRNA,我们还引入了三种类型的sgRNA突变:
- 第一,所有1个核苷酸缺失;
- 第二,所有1个核苷酸缺失。
- 第二,所有1-核苷酸插入;
- 第三,所有的1-核苷酸都与靶DNA错配,
从而产生具有27,897个独特sgRNA的文库。 我们向它们添加了10,618个靶向小鼠Thy1基因座的sgRNA,用作阴性对照。 我们感染了MOLM13细胞,并进行了流式细胞术以分离出CD33阴性细胞(补充图18和补充表18)。
我们用NGG PAM检查了完全匹配的sgRNA的活性,以鉴定导致功能强烈丧失的CD33目标位点,随后的分析仅在该区域检查了sgRNA(补充图19)。使用阴性对照sgRNA定义无活性,我们比较了具有不同PAM序列的sgRNA的活性。不出所料,具有NGG PAM的sgRNA的活性sgRNA比例最高,为91%(图5a)。一些替代的PAM序列导致显着但较小的sgRNA活性比率:NAG(26%),NCG(11%)和NGA(7%)。尽管应避免使用其他PAM来最大化目标上的活动,但必须将其视为潜在的目标外位置。
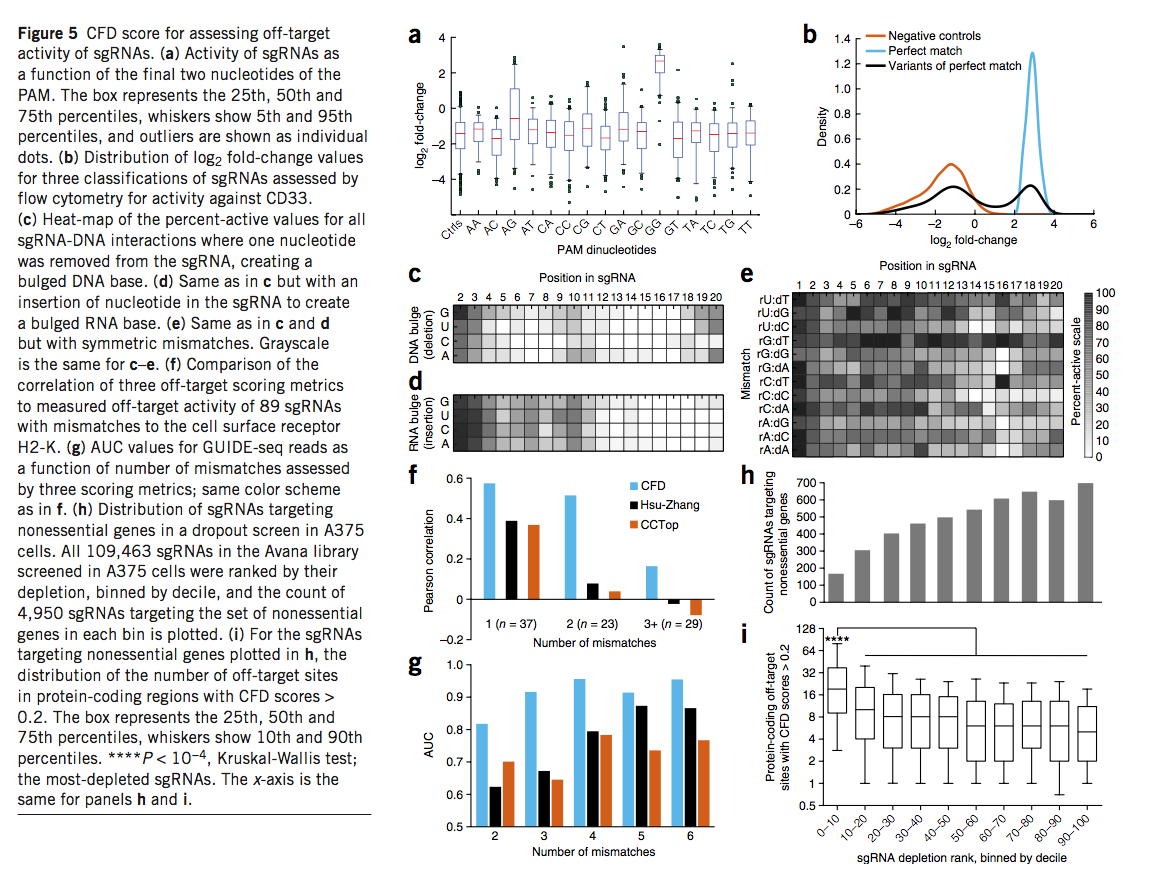
为了检查靶DNA错配,缺失或插入的sgRNA,我们鉴定了65个完全匹配的sgRNA,它们对CD33基因敲除产生了4.8倍以上的富集,持续2 d。高于阴性对照sgRNA的平均值(图5b)及其9,914个相关变体sgRNA。这些变体的活性分布是双峰的,提供了明确的阈值将每个变体归为活跃或非活跃。然后,我们计算了在同一位置共享相同核苷酸突变的sgRNA的活性百分比,即导致活性的单个sgRNA-DNA相互作用的比例,该计算可通过从大量sgRNA采样中获得(补充表19和补充图20)。我们还计算了对数变化倍数(dLFC),以量化每种变体sgRNA相对于其完美匹配sgRNA的活性降低(补充表19)。评估失配变异的两个指标,活性百分比和dLFC,高度相关(R = -0.97,Pearson相关系数,补充图21)。
接下来,我们检查了三种不同类型的变体产生的活动变化。 RNA的缺失很少产生活性(图5c),而RNA的缺失会产生单个凸起的DNA碱基。在76个核苷酸和位置组合中(我们未评估1号核苷酸),使用dLFC度量标准,只有14种此类缺失保留了高水平的裂解(P> 10-4),其中13种发生在2、3位,分别是19和20。值得注意的是,在19和20位,我们观察到对RNA中胞嘧啶核苷的缺失在DNA中产生鼓泡的鸟嘌呤的耐受性较差,只有4%的sgRNA表现出活性,而其他核苷酸的缺失则导致平均有44%的活性sgRNA。同样,sgRNA序列中的额外碱基很少保留活性。只有位置2和3在相同的P值阈值下才产生高水平的活动(图5d)。对于少数例子,已经检查了导致凸出的RNA或DNA碱基的不对称sgRNA-DNA相互作用,我们的结果在质量上是一致的。
最后,我们检查了sgRNA与DNA之间的单碱基错配。总体而言,错配核苷酸的位置和身份在决定活性中起着重要作用(图5e)。例如,即使在PAM近端区域,rG-dT错配也被普遍容忍:在所有位置中,这些相互作用中的256个(289个中的89%)是活跃的。相反,只有37%的rC-dC相互作用保留了sgRNA活性。然而,其他核苷酸错配在不同位置具有完全不同的作用。例如,与DNA中的胸苷有31个错配的30个相比,在位置16的46个嘌呤-嘌呤错配中的0个是有活性的。相比之下,在位置19,41%的嘌呤-嘌呤错配和48%的配对错误的胸苷导致活性sgRNA。这些结果表明,每种类型的错配都必须进行单独评估,并且标出脱靶位点的指标必须结合不完全匹配的RNA-DNA相互作用的位置和特性。
使用这些数据,我们提出了切割频率确定(CFD,cutting frequency determination)得分,以计算sgRNA-DNA相互作用的脱靶潜能。我们测试了89个sgRNA的独立数据集的CFD分数预测脱靶,不完全匹配活性的能力,这些sgRNA旨在靶向H2-D(也称为),其中一些先前已证明可产生有效的蛋白敲除H2-K(也称为),具有高度相似的序列的基因。我们将CFD评分与先前描述的两个脱靶评分指标CCtop42和Hsu-Zhang13评分进行了比较,两者均考虑了RNA和DNA错配的位置和数量,而不是同一性。 CFD评分与这89个脱靶sgRNA的测定活性最佳相关(补充表20)。将数据集除以不匹配的数量,CFD评分在每个子集中表现最佳。如果存在两个或多个不匹配项,则Hsu-Zhang和CCtop得分均与测得的活动相关性较差(图5f)。
为了进一步评估CFD分数,我们检查了使用GUIDE-seq(一种检测sgRNA靶位点的实验方法)获得的结果。在这些数据中,九个sgRNA序列产生了总共402个脱靶位点。我们将GUIDE-seq reads(一种sgRNA活性的量度)与计算出的脱靶评分进行了比较,观察到与CFD评分(R = 0.40)的最佳皮尔逊相关性,其次是CCtop(0.31)和Hsu-Zhang( 0.26)。由于GUIDE-seq是一种表面上无偏的检测离靶位点的方法,因此值得注意的是,只有11个(2.5%)的观察位点获得了CFD评分<0.05,并且是最弱的位点之一,平均reads比完美readds少100倍匹配的sgRNA。
接下来,我们检查了这些脱靶得分指标的特异性。重要的是,我们观察到E-CRISP门户网站使用的对齐工具Bowtie2(参考资料44)不会返回所有可能的不匹配位点,尤其是当不匹配数增加时(补充图22)。因此,E-CRISP和Zhang门户在识别Joung和同事记录的脱靶站点中表现不佳,可能部分原因是无法识别所有高同源性候选站点,而不是一旦找到站点就得分。因此,我们使用了Cas-OFFinder(一种相对较慢但功能全面的工具)来识别与GUIDE-seq检测的九种sgRNA的六种或更少错配的所有可能位点,并使用各种指标对其进行评分。随着潜在的脱靶位点的数目随错配数目的增加而呈指数增加,我们分别进行了检查(不包括一个错配的位点,因为没有这样的位点的zero GUIDE-seq reads,因此无法确定敏感度)。我们观察到CFD评分表现最佳,AUC值在0.82-0.98之间,而Hsu-Zhang评分在0.61至0.87之间,CCtop的评分在0.64至0.77之间(图5g)。我们得出的结论是,CFD评分将能够避免大多数高频脱靶效应。
为了确定脱靶活性对筛选结果的影响,我们使用了精选的927个非必需基因。 Avana库包含针对这些基因的4,950个sgRNA,我们在上述A375细胞的活力筛选中检查了它们的行为。正如预期的那样,这些sgRNA的比例明显减少,显示出强烈耗竭的迹象(图5h)。使用CFD评分,预测出最致命的这些sgRNA的脱靶位点数量在统计学上显着增加(图5i)。该观察结果表明,在负选择筛选中,错误预测的脱靶位点更多的sgRNA经常被错误地消耗掉,因此避免这种混杂的sgRNA可以提高文库性能。
2.4 优化的sgRNA文库:Brunello和Brie
最后,我们将改进的对目标活动的预测(规则集2)与对目标避免的度量标准CFD得分结合在一起,分别为人和小鼠基因组设计了完全优化的文库,命名为Brunello和Brie(补充表21 和22)。 这些新库旨在最大程度地提高规则集2的得分,并最小化具有高CFD得分的脱靶部位(图6a–c)。 因此,Brunello和Brie可能比现有库明显改善。 它们可以通过Addgene获得。 我们的门户网站包含规则集2和CFD模型 ( http://www.broadinstitute.org/rnai/public/analysis-tools/sgrna-design 和 http://research.microsoft.com/en-us/projects/azimuth/ ) ,并提供将这些分数合并到现有工具中的代码(补充代码)。
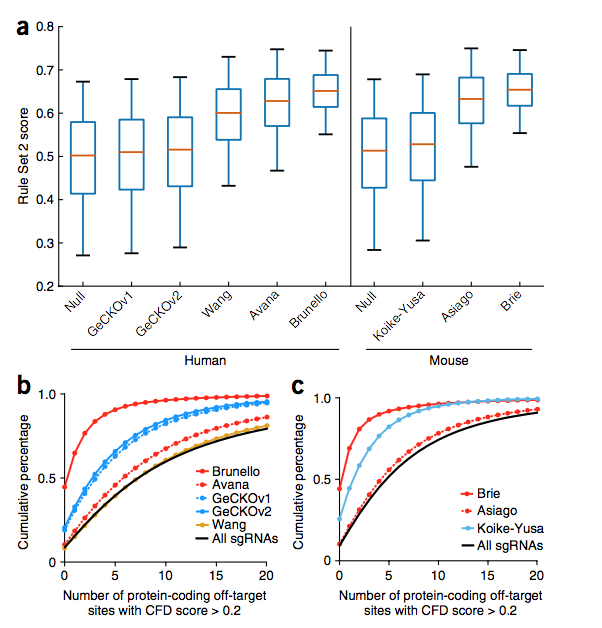
Figure 6 On-target and off-target properties of the Brunello and Brie libraries. (a) Distribution of Rule Set 2 on-target activity scores across libraries. The box represents the 25th, 50th and 75th percentiles, whiskers show 5th and 95th percentiles. (b,c) Cumulative distribution of the number of off-target sites with CFD scores > 0.2 in protein-coding regions across human libraries (b) and mouse libraries (c).
三、讨论
使用先前的合理设计sgRNA的规则(规则集1),我们创建了人类和小鼠全基因组文库并进行了基因筛选。在使用Avana库的正向和负向选择筛选中,预期结果(例如PanCancer基因,core essentials和先前验证的靶标)被多个sgRNA评为强结果,并且与现有文库相比,该筛选产生了更多具有统计意义的基因,从而可以在这些表型分析中鉴定和确认新基因。规则集1对sgRNA活性的预测并不完美,但是,这为文库设计的进一步改进留下了机会。通过将sgRNA活性数据集的大小增加一倍并确定一种更有效的建模方法,我们开发了Rule Set 2,与来自多个实验室的多个数据集相比,Rule Set 1表现出明显的改进。值得注意的是,规则集2预测性能的能力扩展到CRISPRa和CRISPRi筛选。因此,基于规则集2的sgRNA设计(例如我们的Brunello和Brie库)应该在库性能方面带来实质性的额外改进。
决定library performance的另一个主要因素是specificity。 Joung及其同事将他们在实验中检测到的脱靶位点与两种常见的脱靶预测算法所发现的位点进行了比较,并指出“这两个程序都无法识别出GUIDE-seq发现的绝大多数脱靶位点 。尽管规则集1和2代表了数据驱动的靶上sgRNA有效性的定量模型,但缺乏基于类似大规模数据的脱靶相互作用的定量分析,并且无法预测脱靶效应因此相当有限。我们对代表65个DNA靶标的所有单碱基错配和插入缺失的9,914个sgRNA变体进行的分析显示,活性效应的模式很复杂,并且通过检查较小数量的sgRNA-DNA相互作用不会完全显现。我们使用这些数据推导出CFD分数,以预测脱靶切割的可能性,并表明其表现优于其他脱靶得分指标。总之,这些结果为为人类和小鼠基因组创建更具活性和特异性的文库提供了指导。
当前的工作为设计用于大规模筛选和小规模基因编辑实验的改良sgRNA试剂提供了资源。由于筛查面临的最大挑战之一是为感兴趣的生物学方法确定忠实的模型,因此,确认发现的普遍性可能需要检查多种细胞模型。较小的具有较高活性和特异性sgRNA的文库,可通过更好的在靶和脱靶活性预测来进行,这将有助于在一系列模型系统中进行具有成本效益的筛选。直接将CRISPR基因敲除文库与其他基因消除方法(例如最新的RNA干扰(RNAi)和CRISPRi文库)的有效性进行比较,可能会揭示出不同的优势,因此对模型系统的完整评估将受益于多种形式的筛查。此外,未来的工作将确定使用化脓性链球菌Cas9获得的结果是否提供有关其他Cas9蛋白活性的有用课程。这里描述的实验和分析方法说明了一种方法,该方法可发现有助于sgRNA活性和特异性的因素,并针对大规模功能基因组学优化试剂设计。
四、方法
4.1 Avana和Asiago文库
为了设计这些文库,我们靶向由共识编码序列数据库(CCDS,Consensus Coding Sequence Database)注释的蛋白质编码转录本,人类基因组共有18,675个基因,小鼠基因组共有20,077个基因。当一个基因具有多个CCDS ID时,我们就选择每个基因最短的转录本。我们在正链和负链上注释了NGG前间隔物相邻基序(PAM),然后根据三个标准选择了要包含在文库中的sgRNA,并将这些标准分为多个层。最优选的sgRNA将满足所有这三个标准的第一层。但是,并非所有的sgRNA都具有这些特性,因此,要达到每个基因六个sgRNA的配额,就需要跨标准逐步逐步放松层级,并且每轮逐步放松的特性在附录中给出。表1。此外,我们排除了序列中具有BsmBI位点或具有四个或更多胸苷的序列的sgRNA。我们每个基因最多选择六个sgRNA,这导致110,257 sgRNA的人文库(Avana)和120,453 sgRNA的小鼠文库(Asiago)(补充表2和3)。提供了在选择纳入Avana和Asiago库的每一层中这些标准下sgRNA的最终分布。
标准A:目标位置在蛋白质编码序列中的位置,四级除以目标四分位数:(i)蛋白质编码区域的0–25%,(ii)25–50%,(iii)50 –75%,(iv)75–100%。
下表列出了分别选择包含在Avana和Asiago库中的每一层的sgRNA的最终分布:
i; 57%; 57%
ii; 43%; 43%
iii; 0.06%; 0.04%
iv; 0.02%; 0.01%
准则B:to mitigate off-target effects,例如从sgRNA的3’端开始算起的所有长度的靶向蛋白质编码基因的sgRNA,各种长度的序列唯一性,PAM近端(i)13个nt是唯一的, (ii)17个核苷酸是唯一的,(iii)20个核苷酸是唯一的,或(iv)sgRNA序列不是唯一的。
下表列出了分别选择包含在Avana和Asiago库中的每一层的sgRNA的最终分布:
i; 84%; 83%
ii; 13%; 13% iii; 0.2%; 0.4%
iv; 3%; 4%
标准C:规则集1的目标得分,以最大化基因敲除功效,分为十等分; 例如,(i)分数为0.9-1.0,(ii)分数为0.8-0.9,依此类推。
以下列表分别列出了选择包含在每层Avana和Asiago库中的sgRNA的目标得分和最终分布:
i; 0.9–1.0; 3%; 3%
ii; 0.8–0.9; 14%; 15%
iii; 0.7–0.8; 22%; 23%
iv; 0.6–0.7; 23%; 22%
v; 0.5–0.6; 17%; 16%
vi; 0.4–0.5; 10%; 10%
vii; 0.3–0.4; 6%; 6%
viii; 0.2–0.3; 3%; 3%
ix; 0–0.2; 1%; 1%
4.2 文库创建 Library creation
寡核苷酸是在Broad Technology实验室(BTL)的B3合成仪(CustomArray)上合成的。 对于每个sgRNA序列,将BsmBI识别位点与适当的突出序列(带下划线)附加在一起,以克隆到sgRNA表达质粒中。 附加了额外的引物位点,以允许差异扩增同一合成库中的子集。 因此,最终的寡核苷酸序列为:
5′-(正向引物)CGTCTCACACCG(sgRNA,20nt)GTTTCGAGACG(反向引物)。
使用独特的引物组,使用25μl2x NEBnext PCR master mix(New England BioLabs),2μl寡核苷酸池(〜40 ng),5μl终浓度为0.5μM的引物混合物和18μl水扩增单个子池。 PCR循环条件:30sat98°C,30sat53°C,30sat72°C,连续24个循环。
。。。
4.3 Cell culture
。。。
4.4 Virus production
。。。
4.5 Determination of infection conditions for pooled screens
。。。
4.6 Screening
。。。
4.7 Validation of hits
。。。
4.8 Genomic DNA preparation and sequencing
。。。
4.9 STARS
4.10 Development of Rule Set 2
用于目标预测的数据处理。对于每个靶向给定基因的sgRNA,计算了对数的对数倍数变化(LFC, log2 fold-chang),根据read次数判断。接下来,通过为每个基因分配sgRNA的序列,然后将它们重新缩放到0到1之间,从而获得每个基因中sgRNA的归一化等级。因此,给定sgRNA和基因的最终sgRNA得分为[0,1],其中1表示成功敲除。请注意,因为具有更多sgRNA的基因的LFC具有不同的最大值,所以使用归一化等级来代替原始LFC,这表明跨不同基因的LFC不可比。
预测模型。我们在实验中使用了以下统计模型:(i)线性回归,(ii)L1规范化线性回归,(iii)L2规范化线性回归,(iv)先前使用的混合SVM加逻辑回归方法,( v)随机森林,(vi)梯度提升的回归树,(vii)L1逻辑回归(分类器),(viii)SVM分类。这些在python中使用的scikit-learn软件包的实现。对于(ii)和(iii),我们将正则化参数范围设置为搜索在日志空间中均匀分布的100个点,最小为10–6,最大为1.5×105。梯度提升的回归树(学习率为0.1,每个深度估计数为100,最大深度为3)。对于SVM,除非另有说明,否则我们使用具有默认L2正则化的线性内核。
Featurization。核苷酸序列的““one-hot”编码是指采用单个类别变量并将其转换为更多的变量,每个变量的取值可以为0或1,其中最多为“hot,” or on。例如,30聚体目标位点加上背景的位置1可以承担A / C / T / G,并且它转换为四个二进制变量,每个可能的核苷酸一个。这些是“第1阶”功能。对于“第2阶”特征,我们将所有相邻的成对核苷酸视为特征,例如AA / AT / AG / etc。有4×4 = 16个这样的对,因此代表一个这样的对的单个变量被一热编码为16个二进制变量。以前,仅以这种方式使用“位置特异性”核苷酸特征,这意味着对于sgRNA上的每个位置,都使用了一个不同的单热编码特征。但是,在这里,从一些字符串内核文献中获得启发,我们还通过包括“位置无关”功能来扩充这些功能集,例如,对于1阶功能,这意味着一个具有多少A和sgRNA中有许多T等,而忽略了它们的位置,并且类似地出现在2阶(参考51)。因此,对于30聚体(20聚体sgRNA加上背景),我们获得了80个1阶和320阶2阶位置特异性特征,以及4个1阶和16阶2阶位无关特征。 如前所述计算GC计数特征,即,计数20聚体中的G和C的数目,产生一个特征,然后还使用计数> 10的另一特征。
相对于PAM“ NGGN”在N和N位置的两个核苷酸是一键编码的,可产生16个特征,每个NN可能性一个特征(例如AT)。
使用Biopython Tm_staluc函数根据RNA指导序列的DNA版本或其部分的解链温度计算热力学特征。 除了使用整个30-mer靶位点的解链温度和背景,我们还将热力学特征分为三个其他特征,分别对应于sgRNA的三个不同部分的解链温度,特别是五个核苷酸 在靠近PAM的位置,与之相邻的8个核苷酸(远离PAM),然后依次与5个聚体的8个核苷酸相邻(又远离PAM)。
指导位置特征(Guide positional features)(例如氨基酸切割位置和百分比肽)对应于sgRNA切割位点距基因蛋白质编码区起始位置的距离。我们还训练了规则集2的一个版本,该版本不包含与蛋白质信息相关的功能,因为并非所有潜在的靶标都编码蛋白质。
并非所有功能都会出现在补充表17中,因为从所有计算中都删除了完全没有变化的功能。
分层/交叉验证(Stratification/cross-validation)。除非另有说明,否则交叉验证始终是通过一次遗漏一个基因的所有sgRNA来进行的。对于用于设置L1 / L2正则化权重的所需嵌套交叉验证中的内部交叉验证,我们还使用了留一基因外交叉验证。在检查特征重要性时,我们对基因之间的基尼重要性(针对梯度增强回归树)或权重(针对L1正规回归)求平均值。
统计学意义。 当使用完全相同的测试数据和交叉验证时,使用的唯一具有统计意义的检验是比较两种方法之间的Spearman相关性预测量度。为此,我们使用了三组数字:(i)一种方法的Spearman相关性能度量,(ii)另一种方法的Spearman相关性能度量,以及(iii)每种方法的预测之间的Spearman相关。这些值与一种方法一起用于测试相关性的相关度量(此处是相关的,因为每种方法都使用相同的基本事实)。此方法的实现精度导致P值偶尔为0.0。在这种情况下,我们改为报告P <10-16,这是我们观察到的方法报告的最小P值(严格来说,这取决于样本数量以及预测之间的相关性,但这似乎是一个实用的占位符) 。还要注意的是,这种统计显着性的度量并没有赋予每个基因同等的权重,而是每个基因都与其sgRNA的数量成比例地代表。
4.11 脱靶得分
CCtop评分算法的实现如所述。由于CCtop分数与Hsu-Zhang和CFD分数的方向相反,因此CCtop分数的排名沿相反方向进行AUC计算,并且我们报告了所计算的相关值的负值。换句话说,对于CFD分数,值0表示没有预测的脱靶活动,而值1表示完美匹配;相反,低的CCtop得分表明脱靶活动的可能性很高,而高的CCtop得分预示没有脱靶活动。
从Hsu等人发布的数据得出的评分算法。在MIT CRISPR设计服务器( http://crispr.mit.edu/about )上进行了描述。切割频率确定(CFD)得分是使用补充表19中提供的百分比活动值计算得出的。例如,
如果sgRNA与DNA之间的相互作用在第6位具有单个rG-dA错配,则该相互作用得分为0.67。如果存在两个或多个不匹配,则将各个不匹配值相乘。例如,位置7处的rG-dA不匹配加上位置10处的rC-dT不匹配,其CFD得分为0.57×0.87 = 0.50。
4.12 bowtie2性能评估
我们最初尝试使用Bowtie2查找目标外站点。这种方法是成功的,并且相对简单,可以返回所有完美匹配以及与查询序列仅一个不匹配的那些站点。但是,由于Bowtie2算法针对较长的查询序列进行了优化,因此我们无法找到能够可靠地找到多个不匹配位点(例如,许多由GUIDE-seq识别的位点)的选项,这对于我们相对较短的sgRNA查询单个搜索调用中的序列。此外,Bowtie2的运行时性能显着下降,因为我们针对更大的多重失配召回调整了其选项。对于模糊搜索性能较差的工具,一种常见的解决方法是将单个查询序列转换为相关变体列表,对每个变体执行“较少模糊”搜索,并累积结果。利用这种技术,我们得出了“优化的Bowtie2”方法。具体来说,我们对Bowtie2参数空间进行了系统的探索,以找到预排列的数量和类型以及参数设置的组合,这些组合可产生最高的平均召回率,同时在标准计算硬件上仍保持约1 s的搜索时间。这种优化的Bowtie策略可以合理地扩展到全基因组批次,但仍无法识别GUIDE-seq或Cas-OFFinder发现的所有位点。
4.13 GUIDE-seq分析
GUIDE-seq所标识的位点可在该出版物中作为补充表使用。 对于用GUIDE-seq分析的十个sgRNA,Cas-OFFinder被用于寻找在人类基因组中具有多达六个错配的所有脱靶位点。 为了在脱靶评分算法之间进行最公平的比较,我们仅按照CCtop和Hsu等算法的要求,使用NRG PAM对脱靶位点进行了评分,这意味着要排除38个位点。
4.14 Brunello和Brie库的设计
为了设计这些文库,我们以GENCODE数据库( http://www. gencodegenes.org/ )注释的蛋白质编码转录本为靶标。 GENCODE注释是通过将Havana手动注释和Ensembl自动基因注释合并而成的。我们根据GENCODE定义的三个标签为每个基因选择了一个转录本:
- APPRIS( http://appris.bioinfo.cnio.es )批注,该批注根据蛋白质结构信息,功能上重要的残基和证据来对转录本进行优先排序。来自跨物种的比对;
- 转录本的置信度,例如转录本是经过验证的基因座,手动注释的基因座还是自动注释的基因座;
- transcript level support in GENCODE,该支持基于mRNA和EST比对在transcript全长上的匹配程度。
我们注释了针对每个转录本的所有可能的sgRNA,为其“规则集2”评分,并使用这些评分对每个转录本进行sgRNA的靶向活性排名。在此实现中,我们使用了不包含蛋白质靶位点信息的规则集2版本,因为该标准稍后将在选择sgRNA时使用。
同样,我们通过CFD评分各层中潜在的脱靶位点的数量对每个sgRNA进行注释:(i)1.0; (ii)0.2-1.0; (iii)0.05-0.2; (iv)<0.05。然后,我们将每个转录物按(i)层中脱靶位点最少的顺序对每个sgRNA进行排名,并使用其他层作为决胜局。为了打破层次关系,我们对sgRNA进行了优先排序,使其在蛋白质编码区域的脱靶位点最少。 然后以相等的权重组合每个sgRNA的靶上和脱靶等级,从而为靶向特定转录本的每个sgRNA提供最终等级。
为了为每个转录本选择sgRNA,我们首先选择了排名最高的sgRNA,其靶向目标基因的蛋白质编码区域的5–65%。然后,我们从每个转录本中选择了其他sgRNA,从蛋白质编码的角度出发,要求下一个选择的sgRNA靶向的位置与先前选择的sgRNA至少相距5%。这样可以确保目标空间的多样性,这特别有用,因为参考转录本中存在的外显子可能不包含在应用该文库的任何特定细胞模型中。使用这种启发式方法,我们每个基因最多选择4个sgRNA。为了满足某些基因的配额,我们消除了5–65%的蛋白质编码区域和5%的间隔标准。
参考资料:
- Doench,J.G., Fusi,N., Sullender,M., Hegde,M., Vaimberg,E.W., Donovan,K.F., Smith,I., Tothova,Z.,Wilen,C., Orchard,R. et al. (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol., 34, 184–191.
