【5.6.1】基于 RNA 的疗法的 mRNA 结构、稳定性和翻译的组合优化
体外的表达和体内的成正比么?
我们提出了一种大规模平行报告基因检测,称为 mRNA 溶液稳定性和细胞内稳定性和翻译 RNA-seq (PERSIST-seq,Pooled Evaluation of mRNA in-solution Stability, and In-cell Stability and Translation RNA-seq) 的合并评估,它能够系统地确定 UTR、密码子选择和 RNA 结构对 mRNA 的影响 人类细胞中的翻译率和 mRNA 稳定性,无论是在细胞中还是在溶液中。
二、结果
用于系统发现 mRNA 设计规则的组合库
在寻找稳定和高表达 mRNA 的设计规则时,我们旨在表征大量在 5' UTR、CDS 和 3' UTR 区域具有广泛变化的 mRNA 序列设计。我们利用最近商业基因合成的加速发展,开发了大规模并行分析 PERSIST-seq(图 1A,图 S1)。在该方法中,可以平行测定 mRNA 变体的细胞翻译效率、细胞稳定性和溶液稳定性。体外全长转录 (IVT) DNA 模板是通过商业基因合成服务(Twist、Genscript、Codex)获得的。每个模板都包含三个附加功能:(1) 用于执行 IVT 的共享 T7 启动子序列,(2) 3' UTR 中的条形码,以通过廉价的短读测序实现多路复用,以及 (3) 3' 处的恒定区使聚合 PCR 和逆转录 (RT) 反应成为可能的末端(图 S1)。 该文库在体外转录、修饰(3’polyA-tailing 和 5’m 7 G-capping),转染到细胞中并通过池中的条形码测序进行量化(图 1B),通过多核糖体分析或 mRNA 在细胞或溶液中随时间的降解直接测量翻译。
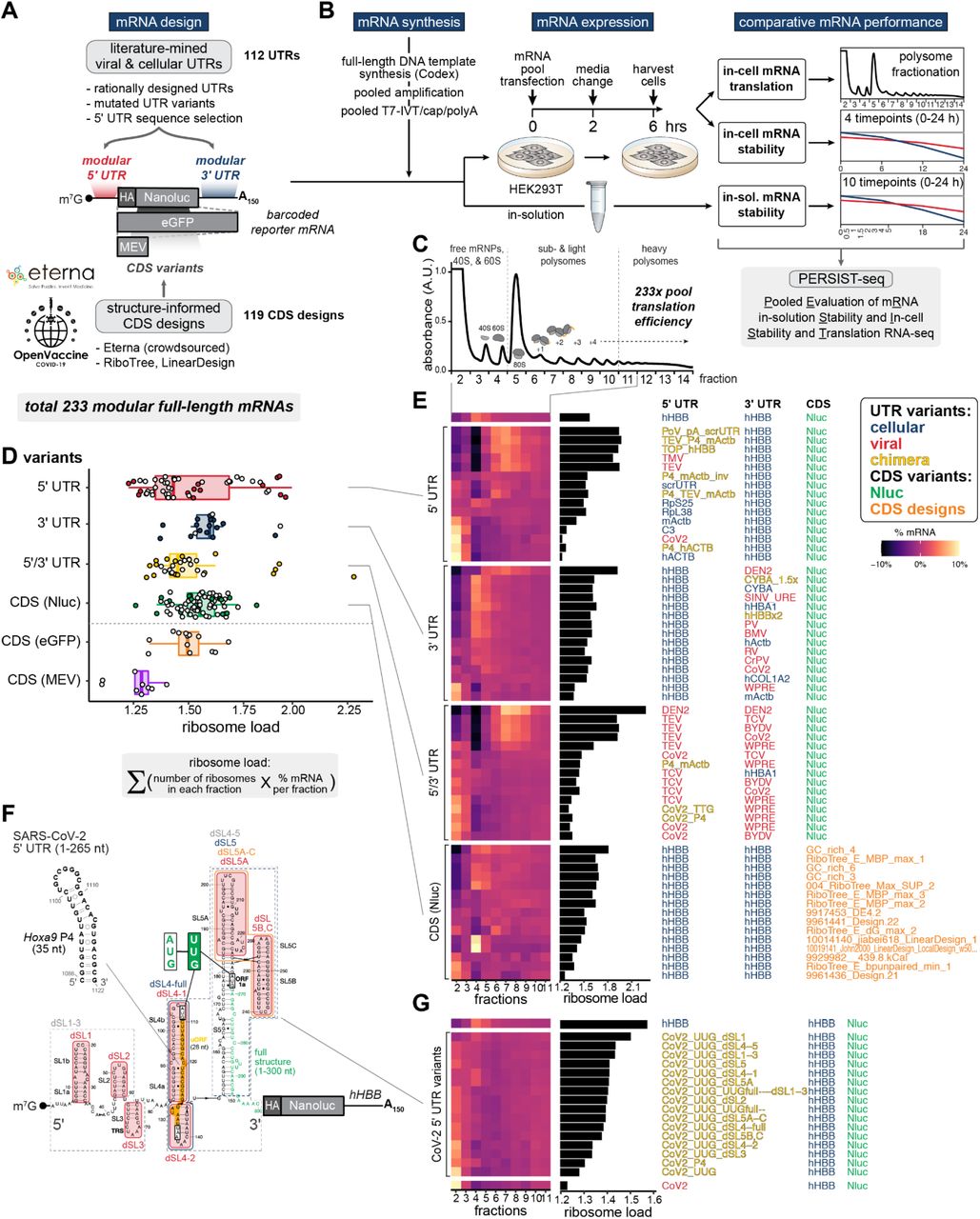
图1 PERSIST-seq overview and illustrative ribosome load insights. PERSIST-seq 概述和说明性核糖体负载见解。 (A) mRNA 优化工作流程概述。文献挖掘和合理设计的 5' 和 3' UTR 与 Eterna 和算法设计的编码序列相结合。然后并行实验测试所有序列的溶液内和细胞内稳定性以及核糖体负载。mRNA 设计在 3' UTR 中包含独特的 6-9 nt 条形码,用于通过短读长测序进行标签计数。 (B) 并行测试溶液内和细胞内稳定性和核糖体负载的实验设计。在转染到 HEK293T 细胞或在溶液中降解之前,mRNA 被体外转录、5' 加帽和聚腺苷酸化。然后收获转染的细胞用于蔗糖梯度分级分离或细胞内降解分析。 (C) 来自具有 233-mRNA 池的转染 HEK293T 细胞的 Polysome 痕迹。 (D) 5' UTR 变体在每个构建体的平均核糖体负载方面表现出更高的差异,这是由多聚体测序确定的。给出了核糖体负荷的公式。 (E) 来自每个设计类别的顶部、中间和底部 5 个 mRNA(按核糖体负载)选择的 mRNA 设计的 polysome 配置文件的热图。 (F) SARS-CoV-2 5' UTR 的二级结构模型。突出显示引入的突变和替换。 (G) SARS-CoV-2 5' UTR 变体多聚体分布的热图,按核糖体负荷排序。

图S1 使用细胞内多核糖体验证的 mRNA 报告基因设计以及细胞内和溶液内工作流程。 (A) 3' UTR 条形码 mRNA 报告器的示意图,用于以混合格式筛选 mRNA 性能。位于可变 3' UTR 侧翼的恒定区和条形码有助于在包含 PERSIST-seq 的每个合并实验中同时扩增和识别数百个构建体。全长 mRNA 的 DNA 模板在 Codex 平台上合成,并使用与可变 5' UTR 之前的恒定区(T7 启动子)和可变 3' 之后的“constant3”区互补的引物在混合 PCR 中扩增UTR。 (B) 从单独合成的 DNA 模板到233 种不同构建体的体外合成 mRNA 池的工作流程摘要。然后,我们使用相同的 mRNA 池在一组三管齐下的细胞内和溶液中表达和稳定性分析中筛选 mRNA 性能。 (C)在体外转录 (IVT) 3 小时后,用溴化乙锭 (EtBr) 染色的 1.2% 甲醛 (FA) 凝胶对 233-mRNA 池进行质量控制。在加帽和聚腺苷酸化之前和之后分析 mRNA 库。合并的 IVT 与起始模板 DNA 池同样有效,无论是否对 DNA 模板池进行 PCR 扩增。显示了对应于三种 CDS 类型的三个主要频段。加载 RiboRuler 高范围 RNA 阶梯 (Thermo Fisher) 以供参考。 (D) 转染的 mRNA 报告基因的 Polysome 分馏分析。例如,在转染到 HEK293T 细胞后 6 小时,具有短乱序5' 和 3' UTR 的mRNA的分布与内源性人类ActB mRNA的分布进行了比较。从馏分中提取 RNA 并通过 qPCR 进行量化,并使用 RNA 掺入进行标准化。将值绘制为按分数标准化的 mRNA。 (E) 含有带有 hHBB 5' 和 3' UTR 的 CDS 变体的条形码 mRNA 的溶液内 RNA 降解策略。CDS 变体的不同降解取决于它们各自的 CDS 结构。mRNA 池在溶液中通过亲核攻击(红色圆圈)降解。降解后,进行 RT-PCR 以选择性扩增沿其全长保持完整的 mRNA。然后,这些全长 mRNA 的条形码区域经过 PCR 扩增、接头连接,并准备用于 Illumina 测序。
5’和3’的设计略。。具体参见文献
我们还包括使用几种算法方法的 CDS:
- 首先,我们包括使用商用算法设计的序列,以优化密码子适应指数 (CAI) 45。
- 其次,我们使用“富含 GC”的方法设计了序列,其中每个密码子都是从 GC 含量最高的密码子中随机采样的,基于 CureVac 研究人员开发的策略9。
- 第三,我们包括使用 LinearDesign 算法设计的 CDS46,它返回一个确定性的最小自由能解,该解由密码子最优性加权。
由 UTR 驱动的高动态翻译范围 High dynamic range of translation driven by UTRs
为了评估与 mRNA 疗法效力相关的翻译效率,PERSIST-seq 将 mRNA 池转染到人类细胞中(此处为 HEK293T)。然后细胞裂解物经历蔗糖梯度分级,将 mRNA 分为主动翻译和非翻译部分,通过条形码区域的 RT-PCR 和 Illumina 测序进行分析。主动翻译的 mRNA 具有更多与其相关的核糖体,并且存在于多核糖体部分中,而非翻译或翻译不佳的 mRNA 存在于游离 mRNA 部分中或与 40S 核糖体亚基相关(图 1C)。在初步研究证实高度翻译的内源性 mRNA 的多核糖体负载差异后,人类ActB,对于转染的对照 mRNA,该 mRNA 具有扰乱(scrambled)短 UTR 序列43(图 S1D),我们进行了 PERSIST-seq 以检查 233x-mRNA 文库中不同构建体的 polysome 谱。我们观察到各个部分的 mRNA 分布存在很大差异(此处表示为核糖体负荷,定义为 mRNA 比例的加权总和乘以一部分中的核糖体数)(图 1D 中的方程)。在 5' UTR 变体组中观察到核糖体负荷的最大变化(图 1D)。这些数据表明,使用不同的 5' UTR 来调整目标 mRNA 的翻译效率具有强大的潜力——比任何其他区域(3' UTR 或 CDS)都要多
我们将上述 UTR 序列的起源分类为“细胞”、“病毒”和“嵌合体”(模块化 UTR 组合)。总体而言,在细胞和病毒来源的 5' UTR 中观察到具有最高核糖体负荷的 mRNA 设计(图 1E,表 S1)。这些5’非翻译区包括:
- 鼠 COL1A2,HOXA9 P4,RPL18A TOP,植物RBCS1A ;
- 痘病毒 poly(A) 前导序列与杂乱的 5' UTR 序列以及植物病毒 TEV 和 TMV 的 5' UTR 融合。
- 登革热病毒 5' 和 3' UTR 都单独增加了核糖体负载,并将它们组合成一个 mRNA 导致了累加效应(图 1E,表 S1)。
所有这些序列都具有比hHBB 5' UTR (1.57)更高的核糖体负载 (1.7-2.3 ),从而确定潜在的 UTR 设计策略以提高 mRNA 的翻译效率。此外,hHBB 5' UTR 与相同 5' UTR 中的 TEV 或 5' TOP 序列等元素的嵌合融合增加了 polysome 负载。总体而言,我们的 polysome 筛选成功识别了广泛的 5' UTR 序列,可以部署这些序列以成功促进细胞内的翻译。最令人惊讶的是,与之前的报告相比,我们发现高度结构化的 5' UTR,例如登革热病毒 (DEN2),可以支持有效的翻译。
。。。。。(举了一个例子,改5’UTR,可以)
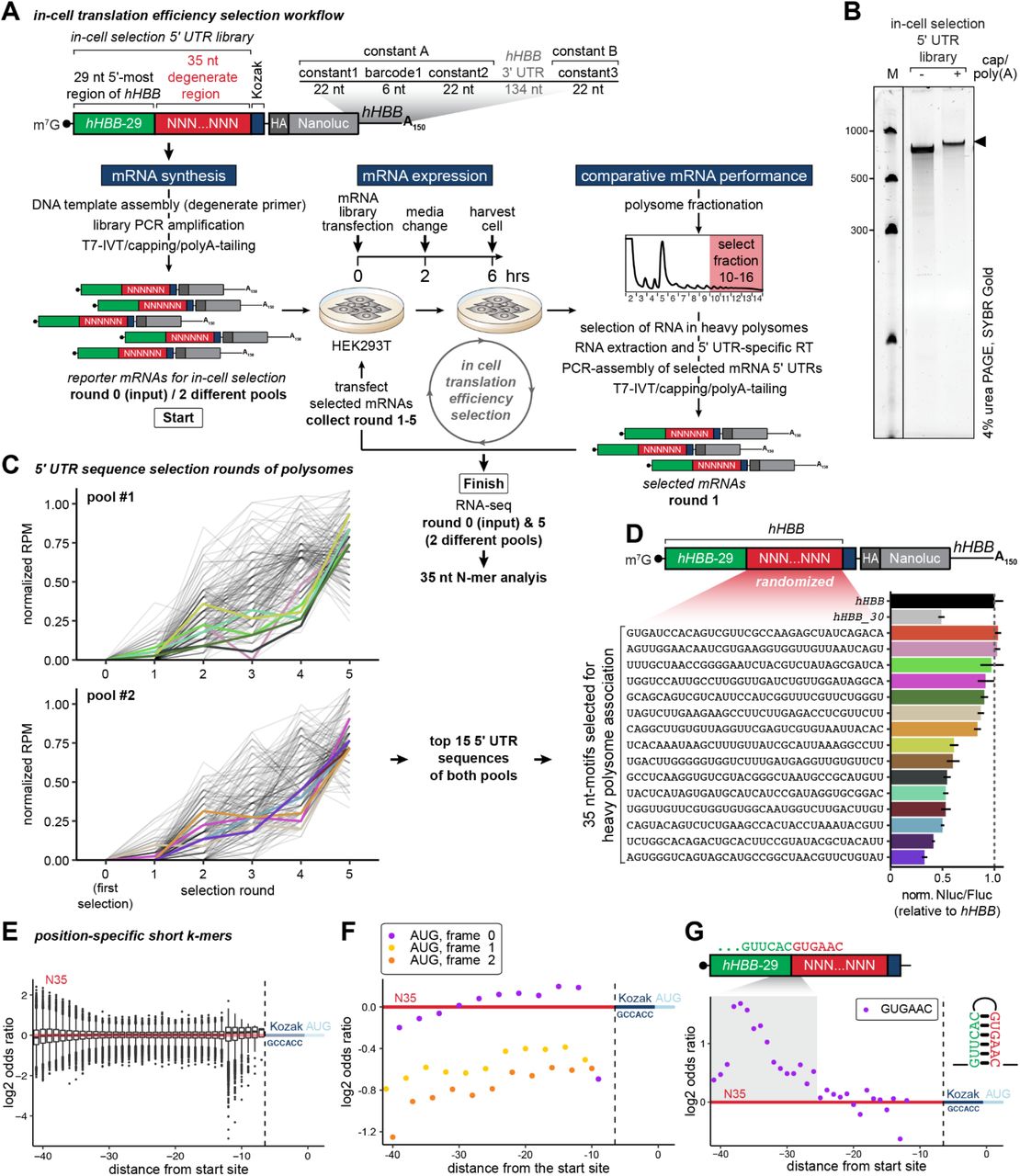
图 S2。 高核糖体加载 mRNA 的顺序选择揭示了有助于蛋白质丰度的 5' UTR 序列。 (A) 旨在揭示有助于翻译效率的 5' UTR 序列的细胞内选择分析概述。选择人类HBB 5' UTR 的前 29 nt作为固定 5' 区域,然后是 35 nt 长的简并区域和恒定的 Kozak 共有序列 (GCCACC)。可变 35 nt 区域的选择是通过随后仅从重多聚体部分纯化的 mRNA 重新转染来引入的。
(B) 变性尿素聚丙烯酰胺凝胶,用于在5' 帽和 polyA 尾之前和之后对体外转录的 mRNA 5' UTR 选择库进行质量控制,显示为选择第 0 轮。所有连续的选择轮产生类似的库。加载低范围 ssRNA 阶梯 (NEB) 以供参考。凝胶用 SYBR Gold (Thermo Fisher) 染色。
(C) 在选择轮过程中,前 5' UTR 序列 (FDR≤0.1) 的每百万 (RPM) 标准化读数。彩色线条表示为荧光素酶报告基因检测选择的 mRNA(总共 15 个来自两个独立的起始池;≥15 的最终轮读取计数,≥2 倍的最终轮富集输入)。
(D) 来自 (C) 的前 15 个 mRNA 的标准化 Nluc/Fluc 荧光素酶活性。多核糖体选定 mRNA 的 5' UTR 中的 35-nt 可变区沿 y 轴列出。它们的荧光素酶活性相对于hHBB绘制在 x 轴上。HBB-29 仅包含 hHBB 5' UTR 的前 29 nt。
(E) 最终 polysome 选择轮与初始起始池之间 k 聚体 (2≤k≤6) 的所有 log2 优势比的箱线图。在 35 nt 可变区的 5'/3' 末端观察到更高的变化。3' 位置的大部分重要 k-mers 是耗尽的。
(F) 在多核糖体选择轮次之后,35 nt 可变区内的框架外(+1 和 +2 帧)AUG 的消耗。帧内 AUG(0 帧)弱耗尽,甚至在 3' 端附近显示出轻微的富集。
(G) 多聚体选择后 6 聚体基序 GUGAAC 的富集。GUGAAC 与 5' UTR (GUUCAC) 的固定 29-nt 区域的 3' 端反向互补。向可变区 5' 末端的富集及其在固定区末端下游第 4 至第 6 个核苷酸的峰值可能表明小茎环结构有利于增加核糖体负载。
。。。。
。结合我们上面的结果,这些 PERSIST-seq 测量表明,与参考 mRNA 序列相比,结构化 CDS 区域和来自细胞和病毒来源的各种 5' UTR 元件可以维持甚至提高细胞内翻译效率。
细胞内 mRNA 稳定性是总蛋白质输出的主要预测因子
mRNA 的总蛋白质输出不仅取决于其细胞内翻译效率,还取决于它转染到细胞后保持完整的时间。为了以混合方式评估构建体文库的细胞内 mRNA 稳定性,PERSIST-seq 量化了在将文库转染到细胞后的多个时间点上剩余的 mRNA 分数。为确保回收完整的全长 mRNA 而不是其降解片段,PERSIST-seq 使用两步法,首先通过逆转录 PCR (RT-PCR) 生成覆盖 mRNA 整个 CDS 区域的扩增子,然后使用侧翼引物只是在短读 Illumina 测序之前进行第二次 PCR 的条形码区域,以计算每个时间点的完整 mRNA(图 S1))。跨时间点的指数衰减函数拟合给出了每个文库构建体的 mRNA 半衰期 (t 1/2 )
对于我们的 233 个 mRNA 文库,PERSIST-seq 提供了广泛的细胞内半衰期动态范围,范围从小于 5 小时到超过 15 小时(图 2A)。我们最初预计由不同的 3' UTR 序列组成的子文库将在细胞内稳定性方面产生最大的变化,因为这些 mRNA 包括多种顺式调节元件,这些元件已知会募集细胞质因子以帮助或防止 mRNA 衰变53 – 56。然而,令人惊讶的是,我们反而观察到 CDS 和 5' UTR 变体组中最广泛的细胞内稳定性变化(图 2A)。我们还注意到具有较高核糖体负载值的构建体往往更不稳定(图 2B)。更具体地说,这些不稳定的 mRNA 包括 5' UTR 或 5'/3' UTR 变体,这些变体是多核糖体转移的(图 2B,多核糖体/单体比率)。与单体的关联增加,因此总体核糖体负荷的更温和增加与 mRNA 稳定性呈正相关(图 2B,单体/前多聚体(前 80S)比率)。重要的是,这些发现确定了 mRNA 设计的一个意想不到的规则——过度提高翻译效率可能会对 mRNA 稳定性产生负面影响。换句话说,随着时间的推移,增加的多核糖体负载似乎与最大限度地表达蛋白质总量的目标适得其反。
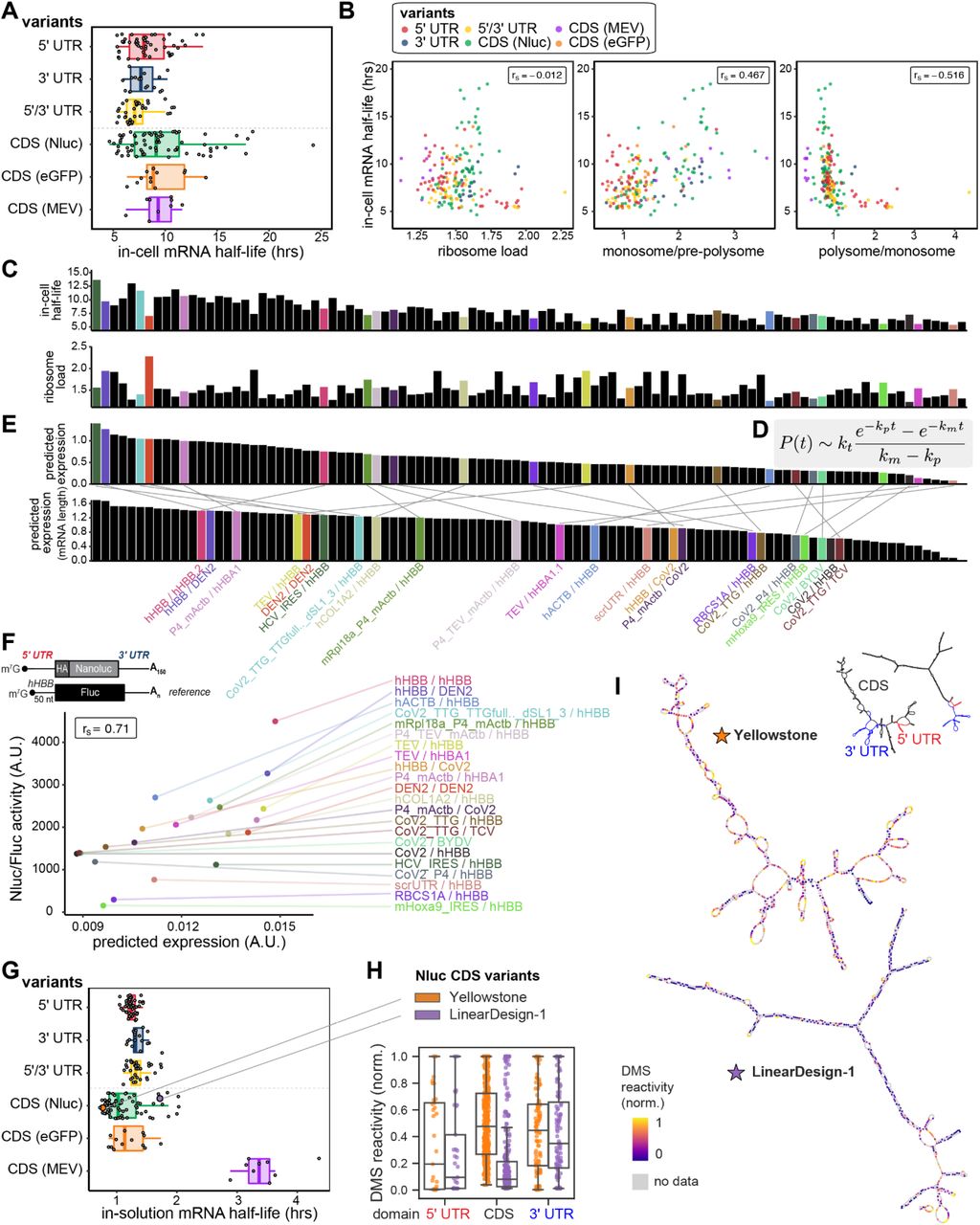
图 2。 细胞内 RNA 稳定性驱动下游蛋白质表达水平。 (A) HEK293T 细胞中每个 mRNA 设计的细胞内半衰期。
(B) 较高的 polysome 负载与减少的细胞内半衰期相关。整个剖面(左)、单体与游离亚基比率(中)或多聚体与单体比率(右)的细胞内半衰期和平均核糖体负载之间的相关性。
(C) 具有不同 UTR 的单个 mRNA 设计的细胞内半衰期和平均核糖体负载。
(D) 用于从 mRNA 半衰期和核糖体负荷预测蛋白质表达的动力学模型。P(t)是时间t 的蛋白质数量;m 0是t= 0 时存在的 mRNA 质量;l是 mRNA 长度;k t为翻译率;和ķ米和ķ p分别为mRNA和蛋白的衰变率,。
(E) 根据 mRNA 半衰期和核糖体负荷,使用 (D) 中的动力学模型预测的蛋白质表达。上:每个 UTR 变体的预测蛋白质表达;注意与细胞内半衰期数据的相似性比(C)中的核糖体负载更相似。底部:通过 mRNA 长度标准化的预测蛋白质表达(对应于每个 mRNA 的等质量转染)。
(F) HEK293T 细胞中预测的蛋白质表达和 Nluc/Fluc 活性的相关性。
(G) 各种 mRNA 设计变体的溶液内半衰期。mRNA 的寿命在很大程度上取决于 mRNA 的长度和设计的结构,这通过加速老化条件(10 mM MgCl 2、50 mM Na-CHES,pH 10.0)下 mRNA 降解的时间进程来揭示。
(H) 核苷酸分辨率体外DMS 映射证实了高度结构化 JEV-HA-Nluc mRNA 构建体“LinearDesign-1”和高度非结构化构建体“Yellowstone”之间的结构可及性存在巨大差异。5' 和 3' UTR ( hHBB ) 在设计之间保持不变。
(I) 从 DMS 指导的结构预测映射到结构的核苷酸 DMS 可访问性。
…………
mRNA 长度和结构驱动溶液中 mRNA 的稳定性
溶液中 RNA 的降解是将 mRNA 疗法分配给患者6的主要障碍。因此,我们对 PERSIST-seq 的最终用途是评估基于结构的策略,以优化 RNA 设计,使其在溶液中更稳定。与细胞内稳定性测量一样,我们测量了降解后保持完整的 mRNA CDS 区域的比例,利用相同的 RT-PCR 选择完整的 mRNA,然后进行 PCR 扩增 3 ' UTR(图 1A、图 S1A、E)。为了模拟脂质纳米颗粒、鱼精蛋白和其他 mRNA 治疗制剂6中可能出现的高效 pH 值和带正电荷的环境,我们使用了含有镁的高 pH 值缓冲液2+加速降解(10 mM MgCl 2 , 50 mM Na-CHES, pH 10.0, 24 °C);在没有 Mg 2+或较低 pH 的条件下,对 RNA 变体的相对稳定性得出了类似的结论(见下文)。溶液中稳定性的结果与 mRNA 文库中细胞内稳定性的结果截然不同。例如,在细胞中,UTR 序列的调节产生了细胞内稳定性的巨大变化(图 2A),这可能是通过影响 mRNA 衰变的细胞机器募集的变化产生的。相比之下,在没有这种细胞机制的水溶液中,改变 UTR 对 CDS 水解降解的 RNA 稳定性产生的影响相对较小(图 2G)。
溶液中稳定性的最大变化发生在 CDS 变体中。先前理论模型7的最强预测是长度变化应该驱动溶液中稳定性的最大变化,不同不同 CDS 类型的 PERSIST-seq 数据证实了 CDS 长度对 RNA 稳定性的影响(图 2G)。池中最短的 mRNA 编码多表位 SARS-CoV-2 疫苗 CDS(MEV),在溶液中的半衰期为 3.4±0.6 小时。编码 eGFP 的最长 mRNA 表现出更短的溶液半衰期,为 1.1±0.08 小时(图 2G),正如预期的那样,潜在水解的位点数量较多。事实上,这些半衰期的比率为 3.0±0.7,在误差范围内与 RT-PCR 捕获的 mRNA 区域长度的反比(958 nt/250 nt = 3.8)相匹配,支持长度效应的理论预测。
溶液中稳定性变化的下一个最大来源是由 mRNA 结构的差异驱动的。在编码单个蛋白质(Nluc、eGFP 或 MEV)的 mRNA 中,溶液中半衰期的差异大于 UTR 变体之间的差异(图 2G),并且这些值与不同的预测指标相关性很好结构(见下文)。在 Nluc mRNA 的 CDS 变体中,溶液中半衰期的最大传播(2.8 倍)。我们在这些具有短半衰期和长半衰期的 mRNA 中选择了两个 Eterna 提交的解决方案进行后续研究:“Yellowstone”,一种使用密码子的设计,模拟了在黄石温泉中发现的生物体中发现的基本频率(高 A/C 含量)58; 和’LinearDesign-1',一种基于LinearDesign mRNA结构优化服务器46的设计。化学结构映射表明,长寿命LinearDesign-1显著更高度结构化比黄石,由硫酸二甲酯(DMS)和选择性2'-羟基酰化与引物延伸(SHAPE)的反应性所评估59,60(图2H,图S3)和由这些数据引导的结构模型(图2I)。总体而言,使用 PERSIST-Seq 对溶液中 RNA 降解的整体评估揭示了 RNA 长度和结构对溶液中 RNA 半衰期的显着影响。
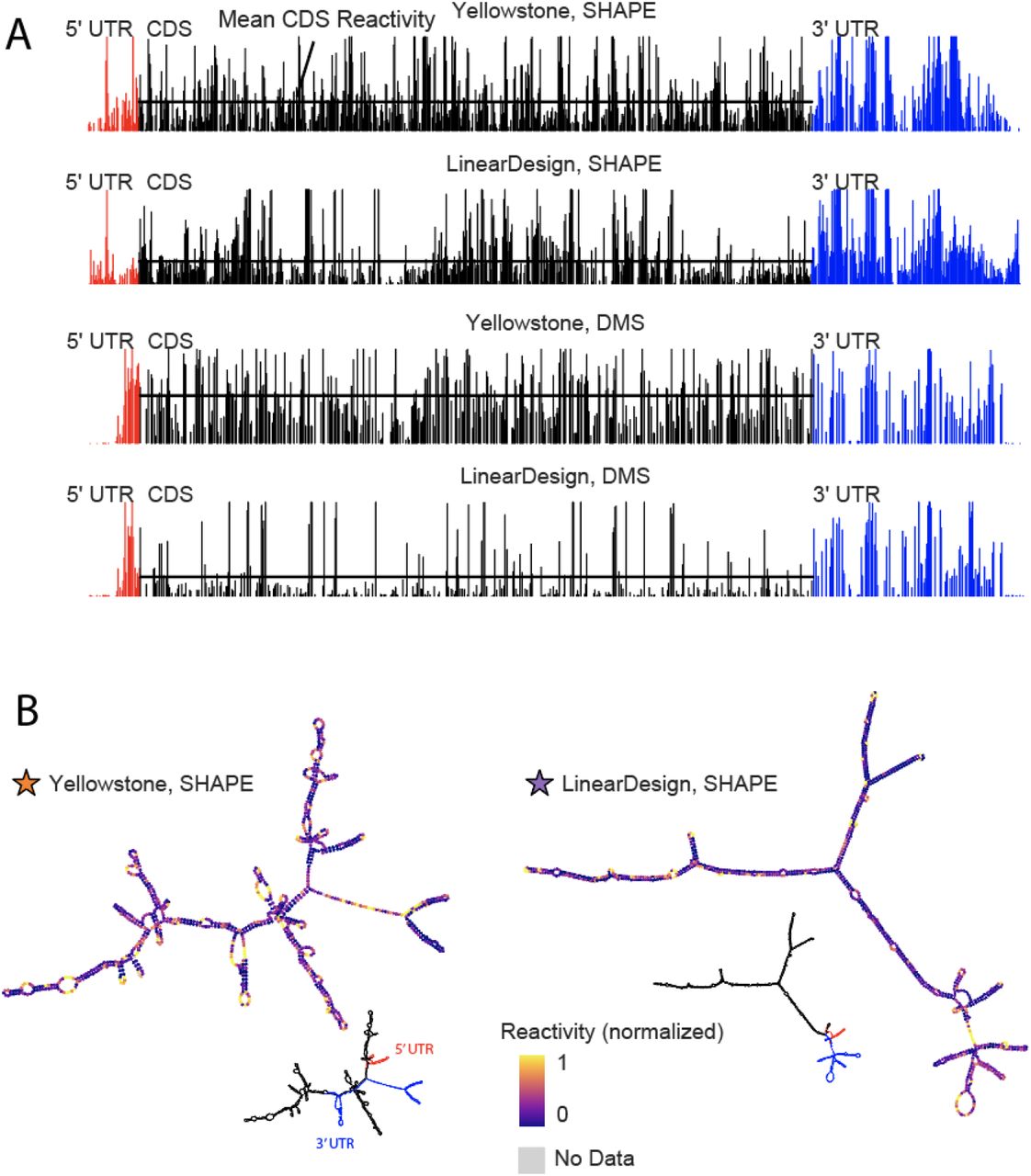
图 S3。 Yellowstone 和 LinearDesign-1 RNA 的化学结构探测。 (A) Yellowstone 和 LinearDesign-1 的每个序列位置的 SHAPE 和 DMS 反应性。
(B) 使用 SHAPE 反应性导出的 MFE 结构。
Eterna-guided In-line-seq 产生额外的设计原则和 DegScore 预测器
上面的 mRNA 设计主要基于 mRNA 结构的计算预测和假设,即结构中不配对的核苷酸将一致地倾向于水解降解,从而改变溶液中的稳定性。我们假设我们可以通过更深入地了解导致此类未配对区域中水解增强或抑制的任何特定序列和结构特征来进一步提高溶液中的稳定性。例如,碱基身份和局部结构特征,如顶端环和内部环的大小和对称性可能在确定溶液中 mRNA 降解61 – 63 中发挥作用。
为了测试这种效果并潜在地发现新的效果,我们要求 Eterna 参与者在一项特殊挑战(“OpenVaccine:Roll-your-own-structure”,RYOS)中生成大量不同的 RNA 分子,这些分子具有设计的二级结构基序。将这些分子的长度限制为 68 个核苷酸并获得独特的 3' 条码发夹,从而能够大规模并行合成和表征数千个 RNA 分子的结构和降解特征(图 3A)。特别是,我们使用 In-line-seq 获得了 3030 个 RNA 片段的单核苷酸分辨率测量值,这是一种低偏差连接和逆转录协议 (MAP-seq) 64在此处适用于在线水解图谱65 (图 3B)。这是第一个将在线探测应用于 RNA 的大规模并行方法和大规模数据集。为了分析序列和结构基序,对序列进行过滤以降低实验噪声,并确保 ViennaRNA 66 中预测的结构与通过同时收集的 SHAPE 映射数据推断的结构相匹配(参见方法);这些过滤器产生了 2165 个序列和相应的二级结构。我们匹配了用于 PERSIST-seq 溶液中稳定性测量的加速降解条件,但也验证了在没有 Mg 2+ 的情况下,在较低 pH 和较高温度下的测量给出了强相关的结果(图 S4A)。在广泛的层面上,数据证实 RNA 结构是在线水解速率的主要预测因子(例如,将 SHAPE 与在线数据进行比较;图 S4A),但仔细观察揭示了额外的序列和结构依赖规则在线水解。
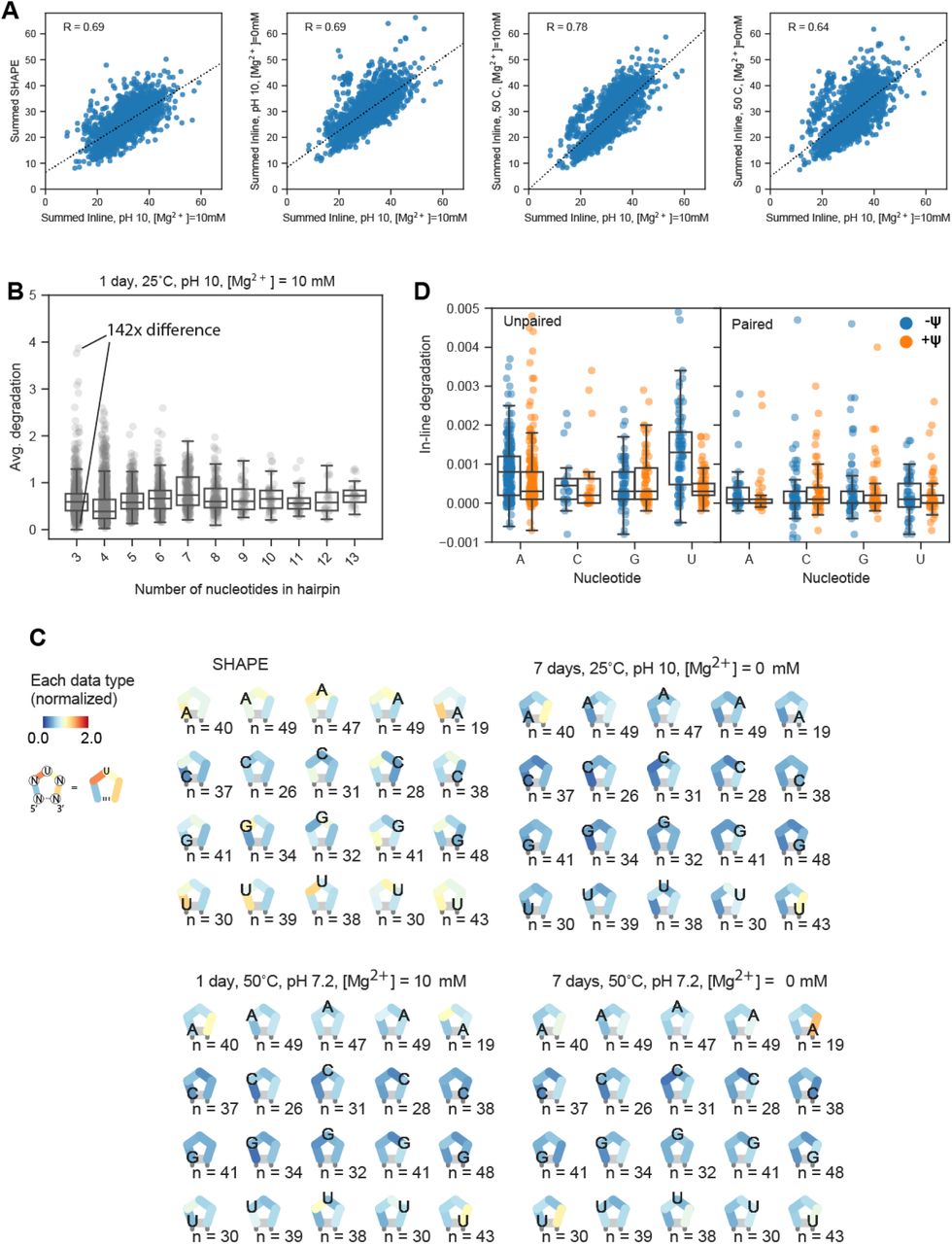
图 S4。 由 In-line-seq 确定的 RNA 降解特征。 (A) 在 pH 10、25°C、[Mg 2+ ] = 10 mM、1 天条件下,每个构建体的总在线降解与所测试的 SHAPE 反应性和其他在线降解条件的相关性。
(B) 在 pH 10、25°C、[Mg 2+ ] = 10 mM、1 天条件下在线降解发夹环降解的平均反应性动态范围。
(C) 三环反应性和降解的序列/位置依赖性对于其他三个实验在线降解条件测试(见图3C)。
(D) 在假尿苷不存在和存在的情况下,用毛细管电泳逐一测量的 8 个构建体的在线降解。左图描绘了预测为未配对的核苷酸,右图描绘了在 ViennaRNA 结构中预测为配对的核苷酸。
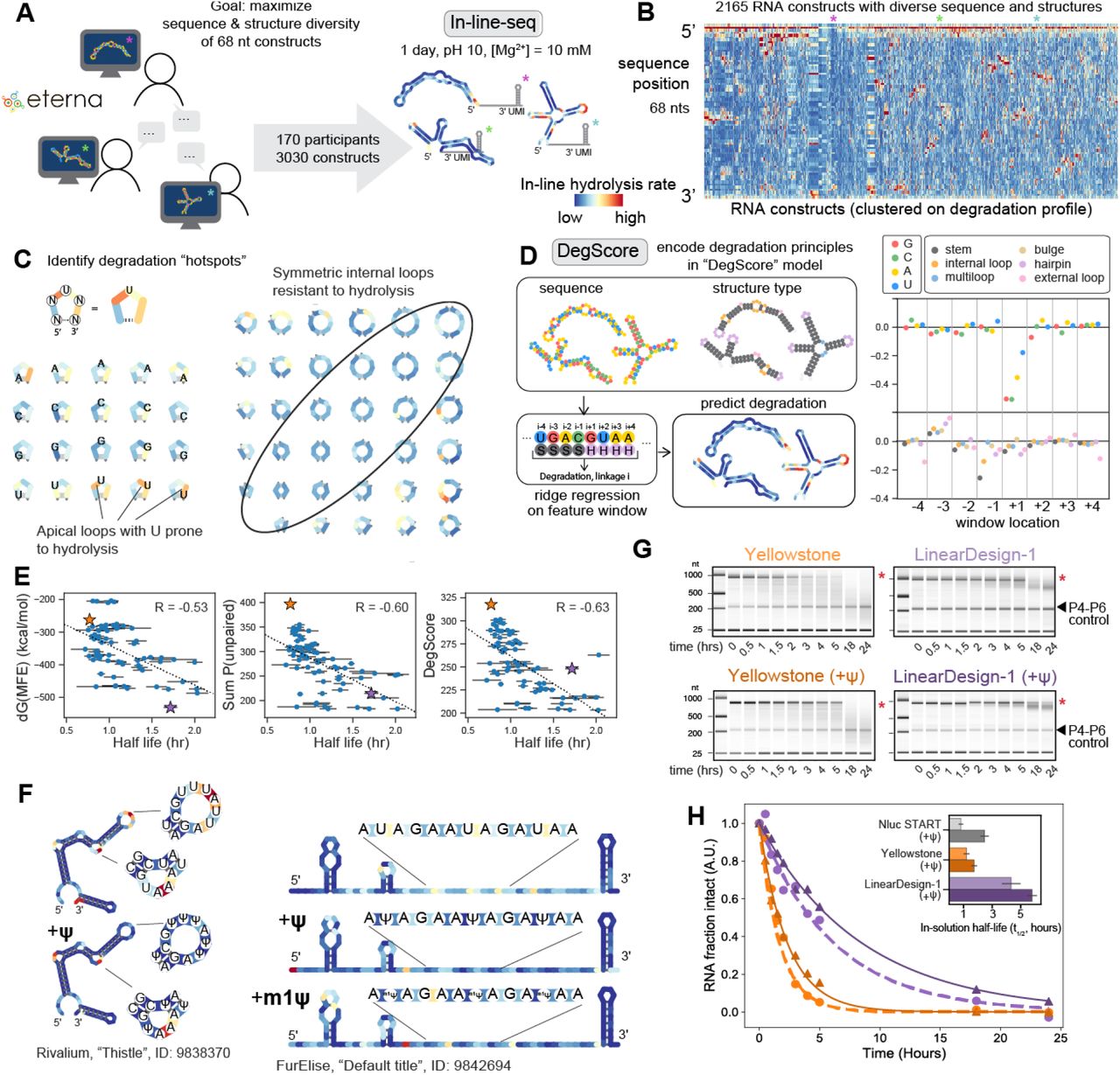
高通量在线水解揭示了溶液中 RNA 降解的原理。 (A) Eterna 参与者被要求设计 68 个核苷酸的 RNA 片段,最大限度地提高序列和结构的多样性。使用高通量在线降解 (In-line-seq) 表征和探测了 3030 个构建体。
(B) 2165 个 68-nt RNA 序列的核苷酸分辨率降解(根据信号质量过滤),由在线序列探测,按降解谱上的层次聚类排序。
(C) 序列跨越一系列不同的二级结构基序,揭示基于序列(即,以 3' 尿苷结尾的连接特别具有反应性)和结构(对称内部环,圈出,与不对称相比抑制水解降解的模式)内部循环)。
(D) 岭回归模型“DegScore”经过训练,可根据序列和环分配信息预测每个核苷酸的降解。具有最大量级的系数对应于链接后立即的序列同一性,U 最不受欢迎。
(E) DegScore 对 mRNA 的预测能力比之前假定的其他两个预测 RNA 稳定性的指标有所提高。
(F) 假尿苷 (Ψ) 修饰的引入稳定了环基序和完全非结构化 RNA 中 U 核苷酸处的选定短 RNA。
(G) Nluc mRNA 分子片段化时间过程的毛细管电泳表征,其设计具有广泛的结构 (LinearDesign-1) 和相对较少的结构 (Yellowstone),使用标准核苷酸和 Ψ 修饰合成。全长 mRNA 带用红色星号表示。的四膜虫核酶P4-P6域的RNA降解作为对照后包括在内。
(H) 十个时间点内完整 RNA 的毛细管电泳测量的指数拟合证实 LinearDesign-1 和 Yellowstone Nluc mRNA 的溶液中寿命之间存在 >3 倍的差异。插图:计算的半衰期。误差棒代表自举指数拟合的标准偏差 (n = 1000)。
当对已知的二级结构基序进行分析时,数据显示给定结构中的 RNA 序列可以显着影响结构基序的降解。例如,在采样最多的三环二级结构的情况下,在线水解速率根据序列变化高达 100 倍(图 S4B)。此外,在许多 RNA 环类型中,似乎导致 3' 尿苷的连接特别容易降解(图 3C,图 S4C),并且在使用毛细管电泳读数的后续实验中重现了这种效果(图.S4D)。因此,独立于 U 的 5' 核苷酸身份,该键是在线亲核攻击的热点63,65 . 此外,我们注意到水解降解的规则取决于核苷酸出现的 RNA 结构环的类型。与不对称内环相比,对称内环中的一个特别显着的特征是抑制水解(图 3C)。为了将这些观察结果提炼成一个预测模型,我们基于这些 In-line-seq 数据(图 3D;参见方法)训练了一个称为“DegScore”的加窗岭回归模型,该模型定量地捕获了诸如链接处水解速率增加等特征,导致3' U(图 3D)。具有最大量值的 DegScore 回归系数对应于连接的核苷酸 3' 的身份(图 3D)。在这些系数中,G 和 C 最有利于具有 3' 连接(水解最少),其次是 A,3' U 对降解最不利,与我们之前的观察结果相符。
为了测试从 In-line-seq 数据得出的 DegScore 指标的准确性,我们对 PERSIST-seq 实验中测量的 mRNA 的溶液内半衰期进行了预测,这些实验是完全独立进行的(图 1B,图.2G)。对于在溶液中半衰期显示出最大差异的 Nluc CDS 变体,我们观察到 DegScore 预测与溶液中半衰期的强相关性(Pearson R = -0.63,p<0.0001)。引人注目的是,DegScore 的准确性优于先前研究中用于参数化 RNA 结构的其他两个指标的准确性,但没有考虑 RNA 水解的序列或结构基序依赖性:预测的最小自由能二级结构的自由能,在几种设计算法中使用的度量,包括 LinearDesign (dG MFE ; R = -0.53),以及预测的 RNA 结构集合7 的未配对概率总和(SUP; R = -0.60)(图 3E)。除了 Nluc CDS 变体之外,我们证实,当对所有测量的 mRNA 进行评估时,DegScore 在预测溶液稳定性方面提供了最高的准确度,包括来自 Moderna 研究人员、Eterna 和 Ribotree 的低和高结构 eGFP mRNA(表 S1和图 S5A) ,乙)。
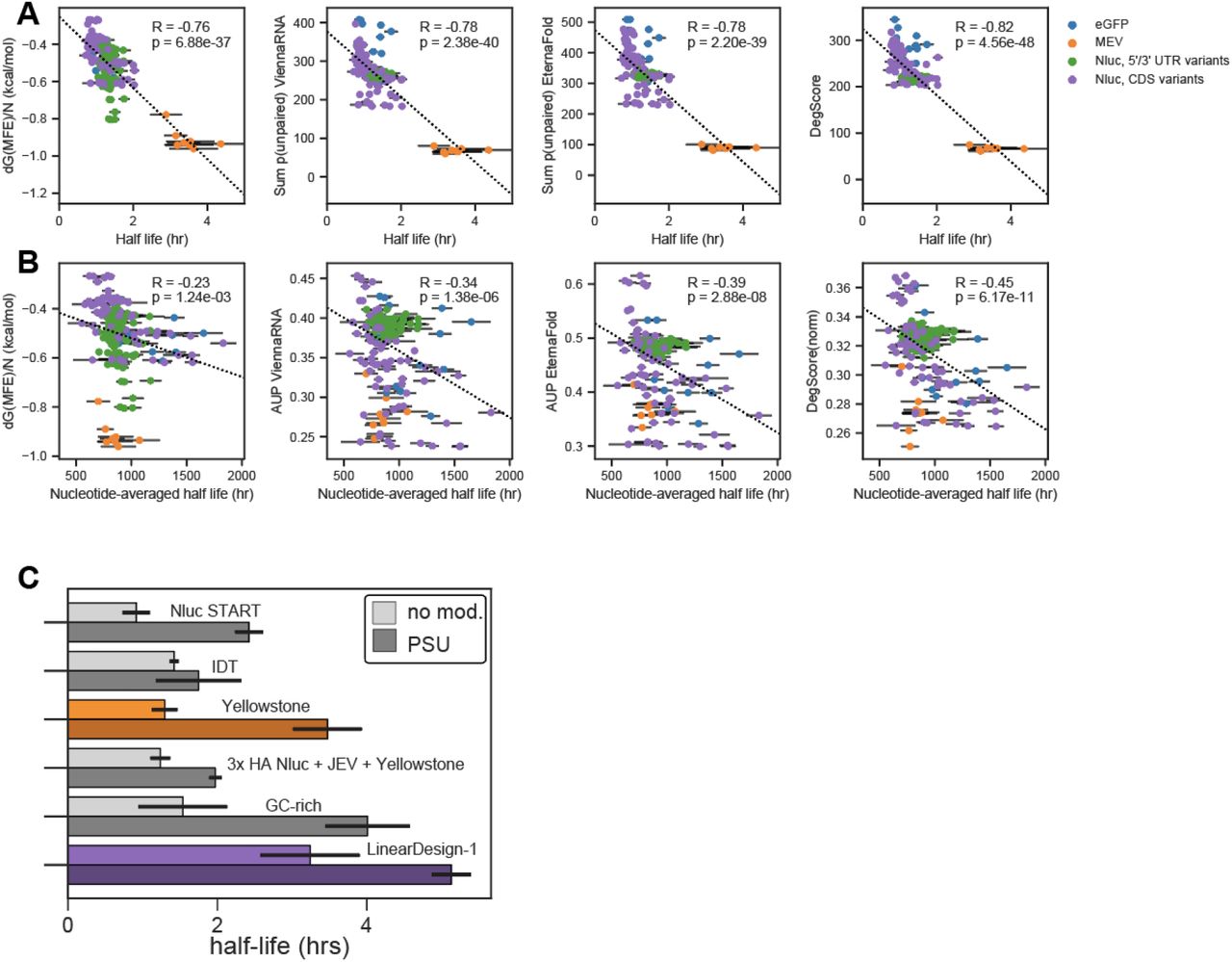
图 S5。 233-mRNA 池中溶液半衰期与 RNA 降解预测因子之间的相关性。 (A) 体外半衰期与 dG(MFE)、ViennaRNA 和 EternaFold 中计算的总和 p(未配对)之间的相关性,以及所有测试模型 mRNA 类型的 DegScore。
(B) 体外半衰期(标准化为 RNA 长度)与 dG(MFE)、ViennaRNA 和 EternaFold 中的平均 p(未配对)(AUP)以及纳米荧光素酶和 eGFP 构建体的 DegScore 之间的相关性。
(C) 6 种模型 mRNA 的体外半衰期的一对一表征,以 U 和假尿苷为特征。
假尿苷稳定溶液中的 RNA
鉴于与 3' U 的连接对降解特别敏感,我们假设 U 的基础化学可能与该核苷的降解能力直接相关,并试图测试 U 的化学替代品是否可以减轻降解。特别是,我们专注于 Ψ 和 m 1 Ψ,因为这些 U 的替代已被广泛用于 mRNA 治疗和疫苗,这是由于细胞内翻译的改进和通过避免更好地控制先天免疫反应2 , 3 , 8 , 67细胞 Toll 样受体(TLR7 和 TLR8)68、RIG-I 和 PKR 69 – 71的识别. 虽然据报道 Ψ 和衍生物可稳定 mRNA 以防止细胞中的衰变67,但尚未报道这些修饰对溶液中 mRNA 稳定性的影响。我们从 Eterna RYOS 挑战中选择了 RNA 序列(图 3A),这些序列被设计为包含富含 U 的环或富含 U 的非结构化区域,用标准核苷酸或用 Ψ 或 m 1 Ψ 代替Ψ 或 m 1 Ψ 重新合成这些 RNA 。 U,并通过毛细管电泳测量它们随时间的在线降解。我们观察到用 Ψ 或 m 1 Ψ取代 U导致在取代残基处显着抑制在线水解,大概是通过改变取代位点的亲核性(图 3F))。我们还观察到在取代的 1 到 2 个位置 5' 处的核苷酸在线水解的抑制(图 3F),可能是由于局部增强的碱基堆积72。基于 SHAPE 和 DMS 分析的结构映射数据证实 Ψ 和 m 1 Ψ 取代没有改变 RNA 的整体结构;在线水解的抑制似乎是由于局部化学或结构效应。
作为对这种出乎意料的稳定作用的进一步测试,我们从 233x-mRNA 文库中制备了六个构建体,包含和不包含 Ψ(图 1,图 S5),包括 LinearDesign-1 和 Yellowstone RNA(图 2I)。使用毛细管电泳测量溶液内降解寿命以评估不同时间点后完整 mRNA 的分数(图 3G-H)。与我们对小 RYOS RNA 的在线水解数据一致,当 U 被 Ψ 均匀取代时,我们观察到这些较长的 Nluc 编码 mRNA 的稳定性提高了 1.2-2.7 倍(图 S5C))。这一发现表明,除了重新设计 RNA 序列以呈现稳定的结构外,还可以通过掺入修饰的 U 核苷进一步显着提高溶液中 RNA 的稳定性。
一对一测试证实了 UTR、CDS 和 Ψ 改进的可加性
到目前为止,
- 我们发现含有高度结构化 5' 和 3' UTR 的 mRNA 可以支持细胞中高水平的蛋白质合成。
- 此外,使用我们的 RNA 降解预测器 DegScore 和 Eterna 衍生设计,我们看到高度结构化的 CDS 可以强烈影响 mRNA 的溶液稳定性及其在细胞中的蛋白质合成(图 2、图 3)。
我们接下来研究了选定的 UTR 和 CDS 是否可以组合以实现稳定且高度翻译的 mRNA,这些 mRNA 从这些单独改进的累加效应中获益。此外,我们研究了 Ψ 代替尿嘧啶的使用,以更彻底地确定其对稳定性和整体翻译输出的影响(图 4)。
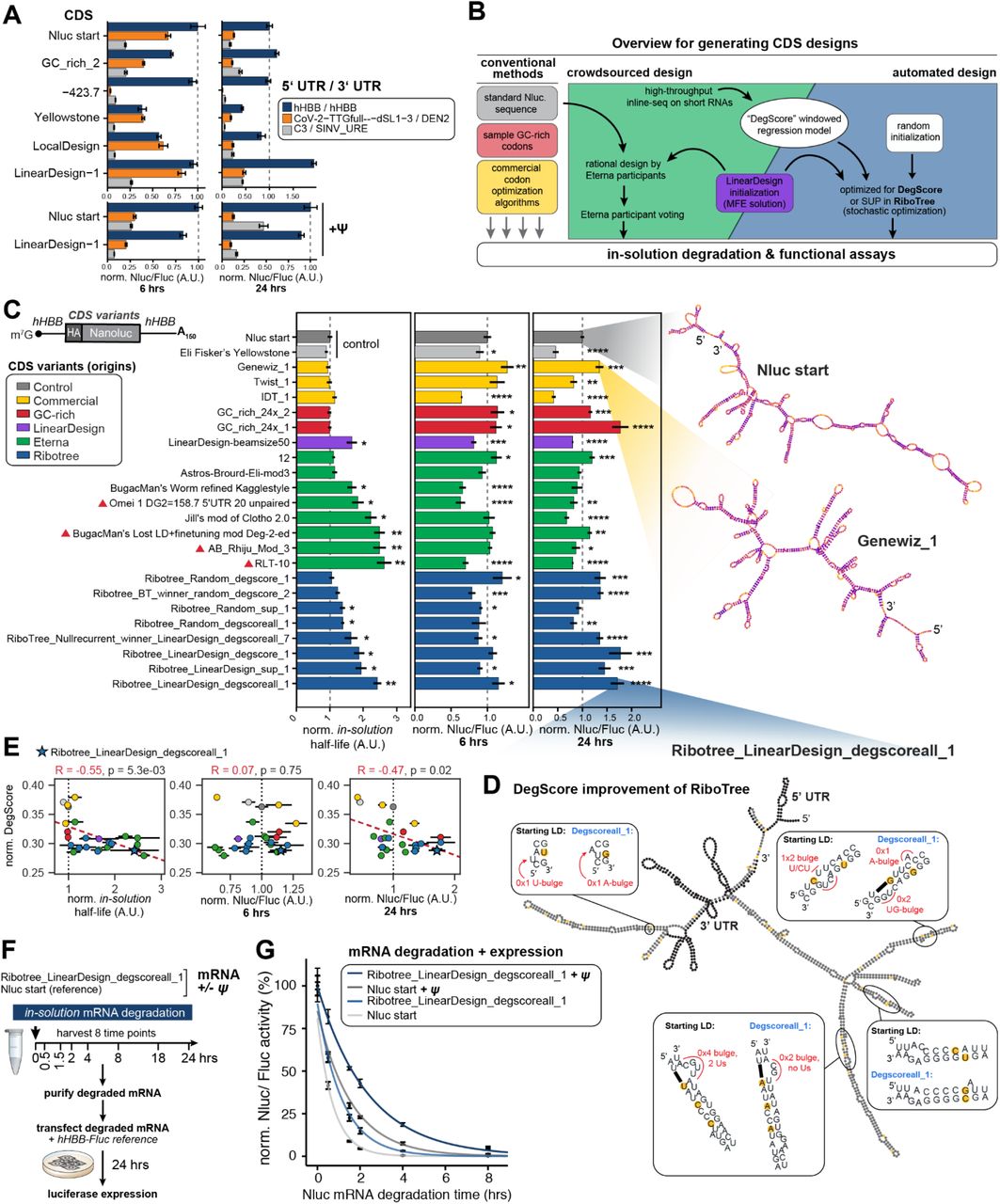
图 4。 5'/3' UTR、结构优化的 CDS 和假尿苷 (Ψ) 的整合一起增强了 mRNA 稳定性和翻译输出。 (A) CDS 和 5'/3’UTR 组合对蛋白质合成的影响不同。体外合成了六个 mRNA 构建体,并在转染后 6 或 24 小时测量荧光素酶活性。在两个选定的构建体上测试了 Ψ 的包含。
(B) 设计 (C) 中测试的 CDS 变体的不同方法的工作流程。
(C) CDS 设计的变化有利于溶液中的高稳定性和差异蛋白表达。体外转录的 mRNA(共 24 个)在溶液中降解或转染到 HEK293T 细胞中 6 和 24 小时。溶液内半衰期和荧光素酶活性被标准化为 Nluc START 参考构建体。显示了所选构建体的预测二级结构,颜色表示每个核苷酸的 DegScore。源自 LinearDesign 解决方案的设计标有紫色三角形。
(D) Ribotree_LinearDesign_degscoreall_1 的预测二级结构概述。放大的框表示 DegScore 对参考 LinearDesign 构造进行的序列优化和后续结构更改。
(E) 在 24 小时而不是 6 小时时增加的溶液半衰期和增强的荧光素酶表达与 DegScore 相关。
(F) 测试 RNA 修饰和 mRNA 设计规则之间对下游稳定性和蛋白质输出的协同作用的示意图。mRNA在有或没有 Ψ 的情况下在体外合成并经受降解条件。随着时间的推移收集样品并在转染到 HEK293T 细胞之前纯化 RNA。转染后24小时测量荧光素酶活性。
(G) 参考 Nluc 序列和 DegScore 优化 CDS 的荧光素酶活性,无论是否存在 Ψ,在溶液中降解后。
我们首先通过从我们的高通量筛选中选择六种 CDS 设计和三种 UTR 组合来确定不同 CDS 设计和 UTR 对 mRNA 稳定性和蛋白质表达的综合贡献(图 1A、图 4A)。选定的 CDS 设计代表了来自不同来源的 CDS,这些 CDS 支持一系列观察到的溶液内半衰期(相对于我们的参考“Nluc 开始”序列,从 0.69 到 1.8 倍;图 2G)。我们将这些 CDS 与不同的 5' 和 3' UTR 组合在一起,这些 UTR 分别被预测和/或确认(图 2F),以促进我们文库中的最高蛋白质表达(图 2E-F)。三种 UTR 组合构成了我们的标准hHBB5' 和 3' UTR;SARS-CoV-2 5' UTR dSL-3 变体与登革热病毒 3' UTR 配对,预计具有高转化效率,但由于长度增加,细胞内半衰期更短;和 C3 5' UTR 与富含 SINV U 的元件 3' UTR 配对,预测具有良好的翻译效率,同时减少侧翼序列的大小(图 2E-F,表 S1)。在溶液稳定性方面,我们预计具有较长 UTR 的 mRNA 在所有 6 个 CDS 中的水解降解半衰期持续降低,毛细管电泳实验证实了这一预期(图 S6)。

图 S6。 UTR 和修饰的核苷对溶液中半衰期的影响。 (A) 6 种选择的 CDS 设计与三对不同的 5' 和 3' UTR 相结合,并测量了溶液中的半衰期。使用 CoV-2-UUG-UUGfull-dSL1-3/DEN2 UTR(红色箭头)的“Nluc start”的半衰期无法准确测量,因为它超出了实验的动态范围;数据代表一个上限。
(B) 图 A 中的两个模型 RNA 是用假尿苷合成的,并测量了溶液中的半衰期。含有假尿苷(红色箭头)的 hHBB/hHBB UTR 的“LinearDesign-1”的半衰期没有被准确捕获,因为该 RNA 持续超出了实验范围;数据反映了一个近似的上限。
误差棒表示半衰期计算的 1000 个自举指数拟合的标准偏差
为了定量评估细胞内蛋白质表达,分别转染 18 种 mRNA,并在 6 小时(作为 mRNA 显着衰减前的翻译率评估)和 24 小时(作为 mRNA 衰减后总蛋白质输出的评估)后测量荧光素酶活性(图4A )。6 小时后,当与hHBB 5' 和 3' UTR组合时,两个 CDS(LinearDesign-1(图 3)和 GCrich_2)与 Nluc start 相比实现了类似的高蛋白质水平。鉴于其在 PERSIST-seq 中不寻常的单体浓缩多核糖体特征,LinearDesign-1 结果引人注目(图 1E)) 以及高 mRNA 结构会对细胞翻译装置产生不利影响的预期。然而,结果与我们的模型一致,即结构化 mRNA 的增强细胞内半衰期可以补偿低翻译效率(图 2C-E)。事实上,到 24 小时时,与 Nluc start 相比,使用 LinearDesign-1 CDS 的 mRNA 显示出荧光素酶产量的两倍增加(图 4A)。LinearDesign-1 CDS mRNA 也表现出特别长的溶液半衰期(图 3G-H)。在测试的三种 UTR 组合和六种 CDS 设计中,大多数证明总体荧光素酶活性低于具有hHBB UTR的参考起始 Nluc (图 4A)。一个例外是 CoV-2-UUG-dSL-3/DEN2 UTR 组合,选择基于其核糖体负载的高性能(图 2D-E),它能够在 6 小时内支持几乎同样高的蛋白质合成水平作为LinearDesign-1 CDS的hHBB UTR;然而,该表达降低了 24 小时(图 4A)。这一发现与与hHBB UTR相比更快的 mRNA 衰减一致,我们的结果支持细胞内 mRNA 稳定性是蛋白质输出的主要驱动因素(图 2C、E)。
鉴于我们观察到假尿苷 (Ψ) 修饰的 RNA 片段和 Nluc mRNA 的细胞内稳定性的改善,我们进一步测试了假尿苷化对 Nluc start 与 LinearDesign-1 CDS 所达到的细胞内稳定性和蛋白质水平的影响(图.4A )。正如预期的那样,用 Ψ 代替 U 制备 mRNA 导致这两种 CDS 的溶液稳定性增加,与 UTR 无关(图 S6)。在蛋白质表达方面,我们观察到在表达 6 小时和 24 小时后,与未修饰的 mRNA 相比,具有固定 CDS 的不同 UTR 组合对总体荧光素酶活性的影响不同。重要的是,带有hHBB UTR 的Nluc start 和 LinearDesign-1 CDS在 6 小时和 24 小时都保持高蛋白表达。图 4A ) 表明,尽管有 Ψ 修饰和高度结构化的 CDS,但翻译仍然持续。总的来说,这些结果证明了 mRNA 稳定性对蛋白质输出的重要性,并表明hHBB UTR、高度结构化的 CDS 和 Ψ 的使用将是提高溶液稳定性和蛋白质输出的最佳选择
所有设计规则的整合导致具有高溶液稳定性和高蛋白质输出的 mRNA
对于我们的最终实验,我们试图测试进一步优化,尤其是 CDS,是否可以增强溶液中 mRNA 的稳定性和细胞内蛋白质表达。为此,我们收集了各种 CDS 设计以与上面的 Nluc start 和 Yellowstone mRNA 进行比较,包括 (
- 可用 mRNA 设计算法的默认输出,包括由 Genewiz、Twist 和 IDT 网站提供的那些可能会增强 mRNA 结构(LinearDesign 46和使用富含 GC 的密码子9),
- 通过额外的 Eterna 竞赛合理设计的高度结构化的构建体(‘OpenVaccine:Focus on the NanoLuciferase mRNA’),以及
- 自动化 mRNA 结构设计工具 Ribotree 的输出,一种可以启动的随机优化算法来自不同的种子序列(随机或线性设计),并在不同预测因子(AUP、DegScore)的指导下优化溶液中水解寿命(图 4B)。
从这些方法中,我们为 Nluc mRNA 生成了 24 种不同的 CDS 设计,并且基于我们上面的研究,我们添加了hHBB 5' 和 3' UTR 作为恒定区以介导高翻译起始率,并在合成过程中使用 Ψ,因为它增强了溶液中 mRNA 稳定性(图 3F-G)
每个 mRNA 设计都经过加速降解和毛细管电泳,以测量每个单独 CDS 的溶液中半衰期。与 Nluc 起始参考序列相比,源自 Eterna 的设计表现出高达 2.6 倍的溶液内半衰期(参见,例如,RLT-10),而来自商业算法或优化 GC 含量的设计往往表现出相似的溶液半衰期(图 4C)。来自 LinearDesign 服务器的设计以及 Eterna 参与者(例如 AB_rhiju_mod3)和 RiboTree 算法对这些设计的修改产生了半衰期比 Nluc 参考序列增加约 2.5 倍的结构(图 4C中用三角形标记的设计))。在测量溶液中半衰期的同时,还分析了单个 mRNA 设计的蛋白质表达。有趣的是,尽管溶液中的半衰期总体上更长,但在 6 小时时,Eterna 和 Ribotree 衍生的设计往往具有与参考 Nluc 序列相似或更低的荧光素酶活性,而大多数供应商衍生和富含 GC 的设计具有轻微的荧光素酶活性。更高的活性(图 4C)。在 24 小时时,与 Nluc 参考 mRNA 相比,测试的 8 种 Eterna 衍生设计中有 6 种荧光素酶活性降低的趋势持续(图 4C,绿色)。然而,相比之下,8 种 RiboTree 优化的 mRNA 设计中有 6 种在 24 小时时表现出比 Nluc 参考 mRNA 更高的荧光素酶活性(图 4C,蓝色)。值得注意的是,RiboTree 输出的序列从 LinearDesign CDS 解决方案开始,在 DegScore 指导下进行优化并考虑到侧翼hHBB UTR 序列,产生了在溶液中都高度稳定的 mRNA(t 1/2 = 2.4,相对于 Nluc start)并在细胞中表现出高水平的蛋白质表达(相对于 Nluc start 增加 1.7 倍)(Ribotree_LinearDesign_degscoreall_1;图 4C)。溶液中稳定性的同时增加以及细胞内蛋白质表达的改善和持续,有力地证明了我们的 mRNA 设计规则的影响。
为了深入了解导致此 RiboTree_LinearDesign_degscoreall_1 序列成功的原因,我们检查了 RiboTree 在计算上最小化初始 LinearDesign 服务器解决方案中的 DegScore 指标时所做的更改。RiboTree 从起始序列修改的特定区域如图 4D 所示(进一步比较在图 S7 中)。这些计算修改的特点是减少循环中 Us 的存在,并移动局部碱基配对以最小化循环的整体大小,即使这种移动会导致额外的更小的循环。这些修改与在 DegScore 预测器中建模的减轻水解一致(图 3D)。综上所述,这些数据表明,通过减少环中 Us 的整体存在和减少发夹数量以产生“线性”高度双链 mRNA,可以提高 mRNA 稳定性和蛋白质表达。
将来自这些最终 12 个 Nluc RNA 的相关数据提供了对影响 mRNA 性能的生物物理和生物物理特征的额外洞察(图 4,图 S8)。最值得注意的是,DegScore 与测量的溶液中半衰期密切相关(R = –0.55,p <0.001),与 24 小时蛋白质表达适度相关(R = –0.47,p = 0.02),但与 6 小时蛋白质无关表达式(图 4E)。然而,6 小时的蛋白质表达与预测的发夹数量(R = 0.70,p <0.001)和“最大阶梯距离73 ”或最大螺旋路径长度(R = -0.55,p <0.001)(图S8C,表S5)。这些观察结果表明,如 DegScore 预测并通过溶液内半衰期量化的对 RNA 水解的抗性对于更长的蛋白质表达很重要,但其他 RNA 序列和结构特征在更短的时间尺度内控制蛋白质表达。例如,更长或更多分支的双链 RNA 茎可能会阻碍核糖体解开 RNA 二级结构的能力。

在上述所有测试中,我们使用结构读数(逆转录 PCR;毛细管电泳)测量了 mRNA 的溶液中半衰期,但在 mRNA 应用中最感兴趣的是降解后保持功能输出。因此,作为最后的实验,我们使用基于细胞中转染和蛋白质生产的功能读数进行了溶液稳定性的实验压力测试。对于 mRNA 功效的这种“端到端”测试,我们合成了原始 Nluc 参考(Nluc 起始)mRNA 以及优化的 Ribotree_LinearDesign_degscoreall_1 mRNA,包括有和没有 Ψ。然后对每个单独的 Nluc mRNA 进行溶液降解,收集 8 个时间点(图 4F)。正如上述表征所预期的那样(图 4C),优化的 Ribotree_LinearDesign_degscoreall_1 mRNA 表现出比 Nluc Start mRNA 更高的溶液内降解抗性,并且将 Ψ 掺入任一 mRNA 进一步增强了溶液中的稳定性,这次是基于细胞中 mRNA 功能的读数(图 4G)。总体差异是显着的:大多数用 Ψ 稳定的 mRNA 和结构优化的 CDS 在加速溶液降解 2 小时后仍保持功能,而我们的起始 mRNA 序列在相同降解后的细胞内活性可以忽略不计(图 4G)。总之,我们对单个 mRNA 的详细研究(图 4) 表明将促进高水平翻译的 UTR 与通过 LinearDesign 和 DegScore 引导的 RiboTree 设计的 CDS 结构优化相结合,我们可以提高 mRNA 的溶液稳定性和细胞内总蛋白输出。此外,下游荧光素酶的表达可以通过 mRNA 的 Ψ 修饰进一步放大。这些结果验证并展示了一个模块化和灵活的平台,该平台可能适用于任何选择的蛋白质靶标,并且可以加速整体改进的 mRNA 治疗解决方案的设计。
三、讨论
基于mRNA的的疗法正在改变,其中人类疾病治疗的方式,特别是感染性疾病(例如及时COVID-19 mRNA的疫苗)74 - 77。然而,要使基于 mRNA 的疗法有效,仍然需要克服几个主要障碍,其中许多与 mRNA 作为分子的内在特征直接相关。
- 特别是,RNA 由于其 2'-羟基而本质上是不稳定的,并且溶液中的 mRNA 通过内嵌亲核攻击降解(可能因配方而加剧)构成了重大挑战6.
- 此外,一旦递送到患者细胞中,候选 mRNA 必须在翻译机制方面胜过其他细胞质 mRNA,并避免细胞 mRNA 降解机制以表达最大量的所需编码蛋白质。
我们的研究以两项新的高通量技术为基础,在应对这两个挑战方面取得了进展。
我们的第一组实验结果源自集成的 PERSIST-seq 技术,该技术能够大规模并行评估 UTR 和 CDS 序列和结构对细胞内 mRNA 翻译效率、细胞内 mRNA 稳定性和溶液中稳定性的影响。该技术将 mRNA 性能的不同可调方面之间以前未知的权衡和累加效应确定为关键决定因素。对于优化这些特征及其相互依赖性最重要的是,我们发现细胞内 mRNA 稳定性可能比高核糖体负载更能驱动蛋白质输出,特别是当蛋白质需要从单剂量 mRNA 长时间表达时。特别是,与最重的多核糖体相关的高翻译效率 mRNA 往往不太稳定:对于最大总蛋白质输出的翻译效率有一个“最佳点”。这种效应可能是由于快速启动导致核糖体在 mRNA 编码区的过度拥挤,这可以诱导空间核糖体碰撞,最近发现这些碰撞导致翻译依赖性 mRNA 衰变78,79。了解这种现象是未来机械研究的一个令人兴奋的前景。总的来说,我们在 PERSIST-seq 中观察到了广泛的 UTR 依赖性翻译效率,可以通过特定的 UTR 序列或 CDS 结构改变进一步微调。我们注意到,对于使用非常短的 mRNA(如用于多表位癌症疫苗,并在我们的 MEV 序列中表示80)或更长的 mRNA(如 SARS-CoV-2 Spike 蛋白等抗原所需要的)的应用,最佳 UTR 和最佳翻译效率的最佳位置可能不同。在这方面,某些 UTR 组合的表现优于hHBB在翻译效率方面,包括多个细胞 5' UTR 以及出乎意料的登革热病毒 5' 和 3' UTR,它们都单独增加核糖体负载,并将它们组合在一个 mRNA 中导致了累加效应。正如 SARS-CoV-2 5' UTR 所示,可以通过旨在缩小病毒前导序列中选择性调节区域的仔细诱变和删除策略来进一步鉴定选择性翻译增强子。我们还注意到,我们的实验仅限于 HEK293T 作为模型人类细胞;高蛋白质输出的最佳 UTR 可能取决于细胞类型,因此会因应用而异。
我们的第二组实验改进与水溶液中的 mRNA 稳定性有关。PERSIST-seq 证实了预测,即高度结构化的 mRNA 的溶液半衰期是常规设计的 mRNA 的两倍多,这对改善溶液中的储存具有重要意义。进一步的设计见解来自一种称为 In-line-seq 的新方法,用于高通量在线探测63 , 65,应用于来自 Eterna 众包平台的数千种不同的短 RNA。在线 seq 结果揭示了许多减轻溶液中 RNA 水解的结构规则以及简单的序列规则:溶液中 RNA 降解的关键决定因素是尿苷的存在以及尿苷 5' 端的 RNA 连接残留物特别容易降解。有趣的是,可以通过包含核苷修饰 Ψ 或 m 1 Ψ来减轻这种影响。带有修饰的 mRNA 的合成——已被广泛用于减轻先天免疫反应和 mRNA 疗法的翻译关闭67 , 68 , 74 , 75 – 因此是一种在维持蛋白质表达的同时显着提高溶液稳定性的简单方法。
进一步利用我们的在线水解数据,我们开发了 DegScore,这是一种水解降解模型,该模型在 PERSIST-seq 数据上进行了独立验证,并在计算机上启用优化任何 RNA 序列。通过将最佳 UTR、DegScore 优化与 RiboTree 和 Ψ 修饰相结合,我们能够实现高 mRNA 稳定性并改善蛋白质表达。我们注意到 DegScore 是针对未修饰核苷酸的降解进行训练的,这并不能解释我们通过 Ψ 观察到的稳定性。因此,未来的研究对来自 Ψ 和其他核苷修饰的降解数据训练类似的模型可能会通过算法设计改进 CDS 的稳定性。我们还注意到,我们的研究侧重于表征裸露的、未配制的 mRNA 的降解,以了解 RNA 的基本生物物理和生化特性对稳定性的限制;对于某些应用,包括个性化癌症 mRNA 疫苗,此类非配制 mRNA 似乎也是最佳选择81 . 对于受益于在脂质纳米颗粒或其他载体中配制的 mRNA 的应用,我们期待未来的研究将 PERSIST-seq 和 In-line-seq 应用于配制的 mRNA 文库,以揭示更多有力的见解。最后,有人提出高度结构化的 mRNA 可以在温度变化、突变和 UTR 变化下保持其结构和溶液中的稳定性,从而激发了术语“超级文件夹”mRNA 7;通过未来的 PERSIST-seq 研究来测试这些预测会很有趣。
总的来说,我们报告了一种新的 mRNA 设计方法,该方法可以显着提高 mRNA 在水溶液中的稳定性,同时维持甚至增加细胞内的蛋白质表达。不必在 mRNA 结构、稳定性和蛋白质输出之间进行权衡。展望未来,我们的计算和实验方法提供了一个平台,可以为新型靶蛋白快速开发定制的高度结构化 mRNA。随着基于 mRNA 的药物被探索用于治疗包括癌症治疗在内的各种人类疾病,我们希望这些见解和方法能够帮助这些药物变得更有效,以更低的每位患者制造成本,更容易获得和广泛分布以缓解疾病。
四、补充说明
细胞内 5' UTR 序列选择以确定有效翻译起始规则
除了比较完整的 UTR 和 CDS 区域之外,我们还试图从复杂的序列库中以无偏的方式选择最佳翻译的 5' UTR 序列(图 S2)。类似于通过直接结合82先前为稳定 mRNA 的 3' UTR 序列而进行的富集序列选择18,我们通过转染具有不同 5' UTR 序列的 mRNA 报告文库并收获与重多核糖体相关的 mRNA 来选择高度翻译的转录本(图 2)。 S2A )。我们通过总共五轮选择和重新转染来自两个独立起始池的重载核糖体的 mRNA,进一步丰富了这些文库,以实现高度翻译的转录本(图 S2A、B)。我们通过 RNA-seq 将它们与初始和第五轮选择的输入序列进行了比较。使用hHBB 5' UTR 作为我们的基线(图 S2A),我们的 5' UTR 文库设计使用hHBB 5' UTR的前 29 nt,然后是一段 35 nt 的随机序列(N35,N=A,C, T,G)和共有的Kozak序列(GCCACC AUG G)83,84的ORF Nluc的上游。
首先,我们询问与hHBB相比,选择与 polysome 相关的 5' UTR 是否会增加蛋白质输出。我们选择了候选 5' UTR,其中我们在最后一轮中观察到了高读取计数(≥15 个读取),增加了所有选择轮的代表性(FDR≤0.1),并且与上一轮选择相比,在最后一轮选择中富集 >2 倍输入(图 S2C)并用这些 mRNA 进行荧光素酶报告基因检测(图 S2D)。除了由候选 5' UTR mRNA 驱动的广泛荧光素酶活性外,我们令人惊讶地观察到,与hHBB相比,没有一个荧光素酶活性显着更高5' UTR。因此,尽管我们选择了最高核糖体负载的 5' UTR 报告基因 mRNA,但这出乎意料地降低了总蛋白质输出,这表明所选的 5' UTR 可能也影响了 mRNA 稳定性或翻译延伸动力学;在正文中描述的 PERSIST-seq 测量中报告了类似的权衡(图 2)。
为了确定所选 5' UTR 序列之间的共同特征,我们使用k pLogo 85(图 S2E)计算了 N35 区域的位置特异性短 k-mer 富集。我们观察到特定 k-mers(总共测试了 165,611 个 k-mers)在 N35 序列的 5' 和 3' 端有更强的富集/消耗(图 S2E)。在从翻译起始的核糖体扫描模型预期的确认观察中,AUG 三联体在 N35 区域显着耗尽(图 S2F)。这种影响是周期性的,并且特定于两个框架外(第 1 帧和第 2 帧)AUG,而框架内 AUG(第 0 帧)没有受到强烈影响,这表明竞争上游起始密码子的负面影响,除非它在-框架以产生 N 端延伸的 ORF 和蛋白质产物。进一步观察到各种其他有趣的基序,例如鸟嘌呤重复序列的消耗(例如 N35 3' 末端的 GGGG 或 GGG 的消耗,接近固定的 Kozak 共识)和整个 5' UTR 的尿苷重复和特定 k-mers 的富集表明形成促进翻译的短茎环结构(表 S3)。后者尤其引人注目:例如,6-mer GUGAAC 在可变 N35-mer 区域的 5' 位置强烈富集;GUGAAC 是固定 HBB-29 区域 (GUUCAC) 最后 6 个核苷酸的反向互补,因此能够彼此完美地碱基配对并包含反向重复(图 S1G)。6 聚体在 HBB-29 区域下游第 4 到第 6 个核苷酸位置的富集峰,因此有利于 3 nt 的插入长度,这将允许在与 6 聚体碱基配对后形成 3 nt 环延伸。检查可变区中其他可能的反向重复 k 聚体作为 6 聚体反向互补沿着固定区域滑动,我们发现茎可能形成到 AUG 的 -30 位置附近。这种模式表明在选择下的 5' UTR 中间的小茎环折叠实际上可能在具有重多核糖体负载的 mRNA 中受到青睐。这一发现与 5' UTR 中二级结构通常抑制翻译起始的典型预期相反。86 . 总之,我们的序列选择策略将先前预测的优化核糖体负载的 5' UTR 序列的规则形式化,并激发了优化蛋白质表达的综合方法,该方法联合利用我们的核糖体负载数据集(图 1)与我们对 in 的研究并行。 -细胞mRNA稳定性(图2)。
五、实验
略
参考资料
- Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics。 https://www.biorxiv.org/content/10.1101/2021.03.29.437587v1.full
