【3.4.5.1】RNA结构与相互作用分析
RNAs在调节其它生物分子和生物过程(例如剪接和翻译)中发挥着重要作用,它们涉及RNA与各种蛋白质和/或其它RNA分子的相互作用。RNA-seq可以用于研究分子内和分子间RNA-RNA的相互作用(RNA-RNA interactions, RRIs),这可能让研究者更好地理解结构组(structurome),或者是研究RNA与蛋白质之间的相互作用,这样就可以深入理解转录与翻译(FIG. 6)。
针对相互作用组(interactome)分析而开发的各种方法都有一个共同的主题:在RNA中富集出那些与其它RNA有相互作用的RNA。一些方法利用的是天然生物学相互作用,而其它的方法则是在目标分子之间计算瞬时作用力或共价键;大多数方法使用的是抗体pull-dwon、亲和纯化或探针杂交的手段来富集RNA进行测序。在这里我们简要描述一下主要的基于RNA-seq的方法来研究结构组和相互作用体的内容。
Figure6–RNA结构和RNA-蛋白质相互作用分析的关键概念
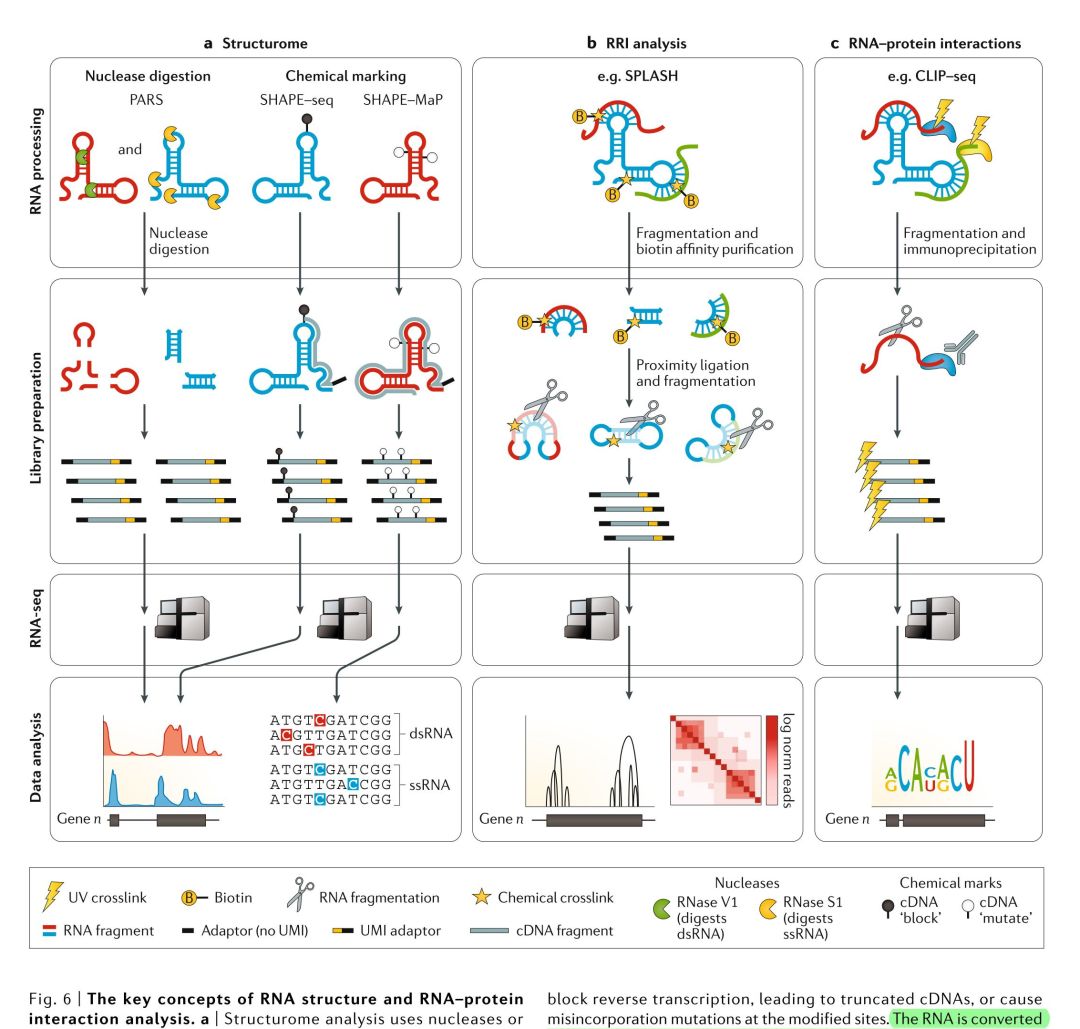
Figure 6-RNA结构和RNA-蛋白质相互作用分析的关键概念。(a)结构组分析使用核酸酶或化学标记试剂在全转录组范围内来研究结构化RNA(例如双链RNA,dsRNA)或非结构化RNA(单链RNA,ssRNA)。在大多数实验中,在单独的反应中对ssRNA和dsRNA进行检测,其结果联合反应性分析法来确定其结构特征。核酸酶消化方法使用针对dsRNA和/或ssRNA的一个或多个核酸酶来研究RNA的结构。例如,在对RNA结构要的并行分析(PARS)中,在体外使用RNase V1(一种dsRNA特异性核酸酶)或S1核酸酶(一种ssRNA特异性核酸酶)来酶切并行样本。酶解后剩余的RNA被转化为cDNA,然后进行测序,测序的读长深度与比对区域的反应性成正比。RNA-seq数据的覆盖和比较结果就能推断RNA的结构。化学分析法(Chemical-mapping methods),例如使用引物延伸的选择性2ʹ-羟基酰化分析法(SHAPE-seq)或突变表达谱分析法(SHAPE-Map),这些方法通过结构依赖形式在体外或体内对双链或单链区域的核糖核苷酸进行修饰。标记物可以阻断逆转录,导至cDNAs的截短,或者是导至修饰位置错误地掺入突变。RNA被转化为cDNA后进行测序,读长深度或突变率与比对区域的反应性成正比,从而推断RNA的结构。(b)RNA-RNA的相互作用分析方法,例如SPLASH,这种方法的第一步是将有相互作用的RNA分子通过生物素化的补骨脂进行交联,然后以通过链霉亲和素对其进行富集,第二步是在邻近位置加入相互作用RNA的自由端加入邻近连接与及片段化。第三步是进行RNA接头的连接以及环化,制备RNA-seq文库用于测序,从而揭示出分子内(也就是结构)的RNA相互作用以及分子间的相互作用位点。(c)RNA-蛋白质相互作用方法,例如RNA交联免疫沉淀后测序(CLIP-seq),这种方法使用UV辐射在相互作用的RNA和蛋白质之间产生共价交联。目的蛋白被抗体富集后,与此蛋白结合的RNA也就被富集了下来,这些RNA加上3’接头后,提取出来用于cDNA的合成。从结合了接头的RNA生成的cDNA用于文库制备,测序。
通过研究RNA分子内的相互作用来研究RNA的结构
核糖体RNA和tRNA构成细胞的大部分RNA。它们与其他结构非编码RNA一起在细胞中发挥各种作用,例如从基因调节到翻译。现存主要有两种研究RNA结构的方法:基于核酸酶的方法和化学探针方法。核糖核酸酶消化于1965年首次用于研究RAN(tRNA(Ala))的结构。在接下来的40年中发展了化学方法,例如,通过引物延伸的选择性2ʹ-羟基酰化法(selective 2ʹ-hydroxyl acylation analysed by primer extension, SHAPE),此种方法用于在单碱基分率水平上检测tRNA(Asp)的结构。但是,只有将各种核酸酶法和化学方法与RNA-seq相结合,才能使方法从单一RNA转移到全转录分析,这正在改变我们对结构复杂性和重要性的理解。在这里,我们集中讨论核酸酶和化学分析方法之间的主要区别(图·6a),如果想对这方面有进一步的理解,可以看Strobel在这方面的综述。
核酸酶方法,例如RNA结构的平行分析法(Parallel Analysis of RNA Structure,PARS)和片段测序法(fragmentation sequencing, FRAG-seq),这两种方法使用能消化单链RNA(ssRNA)或双链RNA(dsRNA)的酶。核酸酶消化后剩余的RNA用作RNA-seq的文库构建。随后通过对产生的RNA序列数据进行计算分析来识别结构化(双链)和非结构化(单链)区域。核酸酶易于使用,可以用于研究ssRNA和dsRNA,但是由于核酸酶消化法的随机特性,它们与化学分析法相比,分辨率比较低。此外,由于核酸酶尺寸比较大,这就限制了这些核酸酶进入细胞,这就使得它们不适合体内研究。
化学分析法使用与RNA分子反应的化学探针,来标记结构化或非结构化核苷酸。这些标记要么阻断逆转录,要么导至cDNA的错配,从而可以定位并分析RNA-seq读长,用于揭示结构组。SHAPE之后进行测序,这种技术方法能够RNA骨架上的核糖2’-羟基反应来标记未配对的ssRNA,虽然发夹环中的碱基折叠会降低其效率。Structure-seq与硫酸二甲酯测序(dimethyl sulfate sequencing, DMS-seq)能使用DMS来标记腺嘌呤和胞嘧啶残基,阻断逆转录,最终从生成的截短cDNAs分析中推断出RNA结构。SHAPE和突变表达谱(SHAPE and utational profiling, SHAPE-Map)和DMS突变表达谱测序(DMS-MaPseq)都修改了实验条件,从而提高了逆转录酶的加工能力,并防止cDNA截短。相反,化学标记会导至错配事件,在RNA-seq数据的分析中,能够检测出这些“突变”,从而揭示RNA结构。化学探针是小分子化合物,尽管由于细胞内的环境处于动态变化中,数据有可能更加多变,但是化学探针还是能够用于研究活体内的有生物学意义的结构。化学探针还可以用于nascent RNAs的结构分析,并揭示共转录RAN折叠的顺序。
核酸酶和反转录阻断方法通常产生短RNA片段,并且只报告单个酶切位点或化学标记,而错误结合和突变检测方法可以报告每个读长的多个化学标记。没有方法不存在偏倚;逆转录阻断永远不会100%有效,本应诱导突变的化学标记可以阻断cDNA合成,这两个因素都可以影响数据的解读。Spike-in控制有可能改善结构组分析的质量,但尚未得到广泛使用。SHAPE方法的比较揭示了仅在体内实验中才会出现效率差异,因此这就突显出比较类似复杂方法时所需要谨慎。
这些方法正在产生关于RNA结构如何在基因和蛋白质调控中发挥作用的新理解。例如,DMS分析说明了,RNA结构有可能调控APA,或许会减慢催化活性区域的翻译,使得更多的时间用于蛋白质的折叠,从而减少错误折叠事件。结构RNA-seq方法的结合有可能产生所有的完整结构组信息。随着该领域的扩展,我们可能会发现,RNA的结构与疾病的进展和或疾病的状态有关;最近的结果表明,异常RNA结构在重复扩张性疾病方面可能发挥作用。最终,结构组分析也许会促进那些靶向作用于研究透彻的RNA结构的小分子的开发,从而开辟治疗开发的新领域。
研究分子间RNA-RNA相互作用
分子间的RRIs在转录后调控中发挥着重要作用,例如miRNA与靶基因的3’UTR结合。现在已经开发了用于研究分子间RRI的工具,它们用于靶向分析和转录组分析。这些分析方法含有一个共同的工作流程,即RNA在打断与邻位连接之前,通过交联来保护其相互作用(FIG. 6b)。大多数并非全部,由不同方法嵌合生成的嵌合cDNA来源于稳定碱基配对(即相互作用)RRNA分子的连接。靶向方法,例如交联,连接和杂交物测序(Crosslinking, ligation and sequencing of hybrids, CLASH), RNA相互作用组分析和测序(RNA interactome analysis and sequencing, RIA-seq)和RNA反义纯化方法测序(RNA antisense purification followed by RNA sequencing, RAP-RNA)能产生一个RNA或RNA家族的高深度相互作用图谱。CLASH丰富了使用IP来进行特定蛋白复合物介导的RRI分析方法,而RIA-seq使用反应寡核苷酸来回收那些与靶基因有相互作用的RNAs;这两种方法都无法区分直接和间接的RRIs,这就导至其生物学解释变得复杂。为了提高RRI分析的分辨率,RAP-RNA使用补骨脂素(psoralen)和其他交联剂,然后用反义寡核苷酸捕获RNA,以及使用高通量RNA-seq来检测直接和间接RRI。虽然该方法可以进行更具体的分析,它需要制备多个文库(每个交联剂一个文库)。
转录组方法从根本上类似于靶向方法:相互作用的RNA在体外被交联后并被富集。通过减少进入连接反应的非相互作用RNA的量来提高富集的特异性,并且可以通过2D凝胶纯化(如在RNA相互作用和结构的补骨脂素分析(psoralen analysis of RNA interactions and structures, PARIS)或交联RNA的生物素亲和纯化(如在补骨脂素交联,连接和选择的杂交测序, sequencing of psoralen crosslinked, ligated and selected hybrids,SPLASH)来实现,或者通过RNase R酶的消化来清除非交联RNA(如在相互作用的RNA连接之后的RNA-seq, ligation of interacting RNA followed by RNA- seq, LIGR-seq)。连接后,在进行RNA-seq文库制备前,去除交联,然后进行测序。PARIS能够生成所有方法中最高数目的相互作用次数,但是每个样本需要75M的读长,这些任何其他的RRI方法都多,并且所需要的DGE实验平均读长深度是其他实验的2倍。
对整理好的RNA相互作用数据的分析可以对多个相互作用进行可视化,并些这种分析方法已经提示了RNA各类的RRI分布的变化。总之,90%的RRIs涉及mRNAs。近一半涉及miRNA或长链非编码RNA,对于这些RNA,大多数相互作用都与mRNA靶基因相关。对这些整理数据的比较揭示了不同方法对特定RNA物种的偏倚,这导至这些方法之间几乎没有重叠。因此,绘制RRI的完整图谱可能需要使用不止一种方法。然而,RRI方法有几个局限性。也许最具挑战性的就是RRI是动态的,并受结构构象和其他分子间相互作用的影响,这使得在没有重复的情况下,很难对其进行解释。分子内的相互作用为分子间的RRI分析增加了干扰,这就需要过滤并除去那些高度结构化的RNAs,例如rRNAs。其它的问题还包括RNA提取过程中相互相互作用的打断,这就需要稳定的交联方法,但最常用的RRI交联剂是补骨脂素和4’-氨基-甲基三氧沙林(4ʹ-amino- methyltrioxsalen, AMT),这些交联剂只交联嘧啶,其效率比较低,会降低灵敏度。此外,邻近连接步骤低效,并且这会连接相互作用和非相互作用RNA,进一步降低灵敏度。
研究RNA-蛋白质相互作用
ChIP-seq已经成了绘制和研究DNA-蛋白质相互作用不可或缺的工具;类似的IP方法也用于研究RNA-蛋白质的相互作用。RNA-蛋白质相互作用方法依赖于IP,利用针对感兴趣的RNA结合蛋白的抗体来捕获其结合的RNA进行分析(第一次报道时是用芯片进行分析的)(FIG. 6c)。各种RNA-蛋白质相互作用方法之间最明显的区别在于相互作用的RNA和蛋白质是否交联以及如何交联:一些方法避免交联(天然IP, native IP),其他方法使用甲醛进行交联,一些方法使用紫外线(UV)光进行交联。最简单的方法就是RNA免疫沉淀测序(RNA immunoprecipitation and sequencing, RIP-seq),时常,但并非所有情况下都使用天然IP法,以及并非总进行RNA打断。这种简便性使用该方法易于被采用。这种方法能产生有用的生物学信息,但是它有两个重要的缺陷。第一,用于保存RNA-蛋白质相互作用的前提是需要进行温和地洗涤,这就意味着富集的片段中有相对高的非特异性结合片段。第二,没有进行RNA打断就降低了结合位点的分析。因此,RIP-seq具有高度灵活性,并依赖于RNA-蛋白质结合的自然稳定性。使用甲醛交联在RNA与其相互作用的蛋白质之间产生可逆的共价键提高了稳定性,并减少了非特异性RNA的回收,但甲醛也会导至蛋白质-蛋白质的交联。这种影响可以通过使用0.1%的甲醛(比ChIP-seq研究使用的甲醛低10倍)进行温和的交联来降低,这能在多个蛋白质靶点上产生高质量的结果。
在CLIP中使用254nm的UV来进行联系是一项关键的技术,它提高了RNA-蛋白质相互作用分析方法的特异性和位置分辨率。UV交联在蛋白质和RNA的相互作用位点产生共价键,但最重要的是,它不对蛋白质-蛋白质相互交联。这就稳定了RNA-蛋白质的结合,允许严格的富集,破坏了天然RNA-蛋白质的相互作用,减少了背景信号。CLIP的实验方法随后就构成了许多方法发展的基础。单个核苷酸分辨率的CLIP(iCLIP)将UMIs整合到文库中,用于移除PCR复制。它还利用了cDNA合成在交联核苷酸处常见的过早截短,通过对截短的cDNA进行扩增来获得交联位点的定量,核苷酸级分辨率图谱。光激活核糖核苷增强片段(Photoactivatable- ribonucleotide-enhanced CLIP,PAR-CLIP)通过使用4 sU和356nM的UV来进行交联。在细胞培养过程中,4 sU被整合到内源RNAs中,356nm的UV辐射会在4 sU整合位点处产生交联(产生高度的特异性)。在产生的测序数据中检测反转录诱导的T>C替换就会能够实现碱基对级的分辨率,并且能够区分交联片段和非交联片段,进一步降低背景信号。最近对CLIP的改进提高了它的效应和灵敏度。红外CLIP(infrared CLIP, irCLIP)用红外凝胶成像技术来代替放射性同位素检验,它是基于珠子的纯化技术。与常规的iCLIP使用的1百万到2百万细胞相比,这些技术的改进可分析只有2万个细胞的RNA-蛋白质相互作用。增强型CLIP(enhanced CLIP, eCLIP)抛弃了RNA-蛋白质复合物的质控和可视化操作,而是在RNA接头中添加了条形码,这种改进可能让所有的样本混合到一起,并用珠子来代替了凝胶。这些改进旨在简化实验操作,eCLIP实验已经研究了近200个蛋白,它已经成了ENCODE项目的一部分。但是,irCLIP与eCLIP目前都没有被广泛采用,部分原因是eCLIP和irCLIP的灵敏性增加的原因是由于其特异性降低导至的,比如利用两个方法所鉴定的PTBP1结合位点上结合或有序和调节外显子的富集减少。随着公共数据库中可用的大量数据为计算分析提供了新的机会,因此谨慎考虑CLIP数据的质控,过滤,以及峰值调用(peak calling)和归一化方法就变得非常重要,这些会影响数据的生物学解释。为了更全面地讨论 RNA-蛋白质的相互作用的CLIP实验方法,我们建议读者可以阅读最近关于这个主题的综述。
一些RRI以及所有的RNA-蛋白质结合方法对IP的依赖限制了其对有良好特征抗体蛋白质的研究,而非特异抗体的结合仍然是一个问题(虽然这一问题并非局限于这个领域)。RNA结构也会影响RNA-蛋白质之间的相互作用;一些蛋白质能识别特异的RNA二级结构或与这些结构竞争结合RNA,这使得体外的发现转向体内就变得复杂了。此外,结构和RNA-蛋白质相互作用方法通常报告一个特定转录本或位置的平均值。在实验室方法中,在计算方法和单分子测序方面的未来发展或许有助于破译一些这些生物变异。
结论
Wang,Gerstein和Snyder关于RNA-seq将“革命性地[如何]分析真核转录体”的预测肯定是正确的。但是,即使是他们,也有可能对这种转型的规模感到惊讶。现在我们可以分析RNA生物学的许多方面,这对于基因组功能、研究开发和确定导至癌症和其他疾病的分子调控异常方面来说是必不可少的。虽然生物学发现阶段还远未结束,但是已经在临床中使用了RNA-seq方法。单细胞测序正在成为许多实验的标准配置,空间转录组学的分析可能会遵循类似的路径,使其能够在与开发当前方法的实验室范围之外使用。长读长测序方法也有可能取代当前相当大比例的研究者们默认选择的Illumina的短读长RNA-seq。对于这种情况的出现,长读长测序技术还需要在增加通量和降低错误率方面做出极大的改进。然而,长读长mRNA异构体测序的优点是,如果它变得像现在短读长测序一样便宜和可靠,那么对于那些除了易降解材料外,长读长测序就可能是首选。考虑到这些因素,那么任何关于RNA-seq在未来十年可能如何发展的预测都有可能过于保守。
参考资料
