【6.6】具有工程RNA二级结构的CRISPR系统的特异性增加
CRISPR(聚类的规则间隔的短回文重复序列)系统已广泛用于基础科学,生物技术以及基因和细胞疗法。在某些情况下,这些细菌核酸酶表现出脱靶活性。这对治疗应用造成了潜在的危害,并可能混淆生物学研究的结果。因此,提高这些核酸酶的精确度受到广泛关注。在这里,我们显示,将发夹二级结构改造到单个向导RNA(hp-sgRNA)的间隔区上,与各种CRISPR效应子结合后,可以将特异性提高几个数量级。我们首先证明,设计的hp-sgRNAs可以基于化脓性链球菌(SpCas9)的Cas9调节反式激活因子的活性。然后,我们显示hp-sgRNA使用5种不同的Cas9或Cas12a变体提高了基因编辑的特异性。我们的结果表明,RNA二级结构是可以调节多种CRISPR系统活性的基本参数。
一、前言
CRISPR–Cas系统是细菌和古细菌中的适应性免疫系统,并且已被证明具有强大的基因组编辑功能。 由于其简单性,将CRISPR-Cas系统用于基因组编辑的努力主要集中在2类CRISPR系统上。
- 第1类系统使用多蛋白复合物靶向核酸,
- 而第2类系统使用称为Cas效应子的单个Cas蛋白,可以很容易地对其进行重组和利用,以用于各种应用。
病毒和原核生物之间的军备竞赛推动了Cas效应子的巨大遗传多样性。每个Cas效应子都有其独特的特性(例如,核酸偏好性,与原间隔子相邻的基序(protospacer-adjacent motif,PAM)要求,Cas效应子的大小),使其具有特定应用程序的优缺点。因此,对2类CRISPR系统的鉴定和表征是一个活跃的研究领域,其首要目标是找到具有新颖或改良特性的Cas效应子。自从SpCas9的最初表征以来,在哺乳动物细胞中活跃的Cas效应子的数量已经扩大到包括:II型CRISPR系统的紧凑型Cas9效应子,V型系统的富含(A + T)的PAM和靶向RNA的Cas13变体的Cas12a(以前为Cpf1)效应子。
尽管这些核酸酶是在其天然环境之外进行基因编辑的通用工具,但它们也具有脱靶效应,导致与间隔子序列不完全互补的位点上意外的DNA断裂。因此,提高这些核酸酶的特异性是一个关键目标,特别是对于基因治疗应用而言。通过合理设计提高2类CRISPR系统特异性的方法主要集中在SpCas9上,并采取了两种通用策略。
- 第一个策略是创建一个AND gate,该门需要两个Cas9分子的协调结合,对核酸酶活性提出了更严格的要求。
- 第二种策略是通过Cas9单向导RNA(sgRNA)复合物减少DNA interrogation的能量,这会导致特异性的整体提高。
第二种策略特别有吸引力,因为与第一种策略不同,它不会增加基因编辑系统的组件数量。这简化了基因传递,这通常是关键的障碍。尽管先前使用这两种策略的努力都取得了成功,但它们受到多种限制中的一个或多个限制,包括与病毒包装限制条件不兼容,系统中的组件数量更多以及需要进行大量蛋白质工程。最近采用定向进化(directed evolution)而不是合理设计的研究产生了许多具有改进特性的新变体。然而,有待观察的是,这许多方法中的哪一种将在整个CRISPR系统中具有普遍适用性。因此,需要一种简单的方法来增加多种CRISPR系统的特异性。
采用合理的设计并采用第二种策略,我们假设对sgRNA进行工程改造可能是调节各种CRISPR系统的一种手段。具体来说,
- 我们通过在sgRNA(hp-sgRNA)5’端延伸设计的发夹(hairpin),将RNA二级结构工程化到间隔子上。
- 然后,所得的发夹结构可以用作R-loop 形成的空间和能量屏障。
- 我们假设通过调节二级结构的强度,R环的形成可以在靶位上完成,但在离靶位上可能受阻,这由于RNA-DNA错配而降低了能量。因为R环的形成是控制SpCas9构象变化为活性核酸酶的关键过程,[29,30]因此,这会阻断脱靶核酸酶的活性并导致特异性增加。
- 由于CRISPR核酸内切酶在其结合通道内容纳了一个核酸双链体,因此我们假设也可以容纳hp-sgRNA的RNA-RNA双链体,而不会干扰sgRNA-蛋白质复合物的形成。
- 此外,hp-sgRNA的设计和生产都很简单:RNA发夹通常遵循Watson-Crick碱基配对准则,而sgRNA的生产方法既快速又便宜。
二、结果
2.1 hp-sgRNA的设计注意事项。
RNA可以折叠成许多不同的复杂结构。对于我们最初的工程结构,我们采用了RNA发夹,RNA发夹是许多RNA分子中的基本结构单元。 RNA发夹由茎和环两部分组成,我们通过扩展间隔区的PAM远端产生hp-sgRNA来创建它们(图1a)。所有设计均通过计算机内结构测定来告知,并且仅将间隔序列用于这些预测(即,排除tracrRNA或crRNA中的结构序列)。

我们预期二级结构的热力学稳定性是hp-sgRNA的重要特征。但是,有很多变量可以用来创建具有相似稳定性的不同结构(图1a)。茎可以沿着20个核苷酸间隔区的任何区域放置,这可能会对R环形成动力学产生可变影响。茎长度(发夹稳定性的主要决定因素)也可以变化。为了调节稳定性,但不一定调节总体hp-sgRNA的结构,可以用非规范性rG-rU碱基对替代茎中潜在的rG-rC / rA-rU位点。自然界中发现的许多RNA发夹都利用5'-ANYA-3’或5'-UNCG-3’四环,它们具有良好的碱基堆积行为。我们将这些四环用于我们的初始结构,但也可以将一部分间隔子本身用于发夹环。在这项研究中,所有这些变量均用于生成hp-sgRNA。此外,为了控制sgRNA长度的任何影响,我们还设计了非结构化sgRNA(ns-sgRNA),其具有延伸到间隔区的功能,但其延伸预计不会形成任何二级结构。
2.2 hp-sgRNA调节基于SpCas9的转录激活因子
我们首先测试了预测的hp-sgRNA结构对Cas9与DNA结合的影响。至关重要的是,我们想分析人类细胞中的这种相互作用,其中有报告显示,可以将sgRNA 5’端的延伸序列加工回天然间隔子的长度[19,33]。因此,我们决定利用无核酸酶的基于dCas9的转录激活因子,其中内源基因激活可以作为dCas9与靶标DNA结合的敏感手段。
对于我们最初的hp-sgRNA设计,我们使用20核苷酸间隔子外部的四环,并使用规范的Watson-Crick碱基配对将发夹茎置于间隔子的PAM远端。我们使用了针对IL1RN内源性启动子的间隔子,IL1RN是我们先前已经高效激活的基因。将sgRNA变体和dCas9-P300反式激活子转染到人细胞中,我们观察到hp-sgRNA可以调节目标基因座上的基因激活(图1b),表明dCas9结合的调节。
我们观察到了hp-sgRNA间隔区延伸的长度与对dCas9结合的影响之间的规律关系(图1b)。观察到唯一的不规则现象是hp15,它具有未配对的5’鸟嘌呤,这是U6启动子所必需的。重新绘制每个hp-sgRNA变体的活性,作为其预测结构的热力学稳定性的函数,我们观察到基因激活在四个数量级上单调下降(图1c)。这些数据提供了预测的RNA结构在人细胞中形成的证据,并证明了计算机预测的结构自由能是其对dCas9与基因组DNA(gDNA)靶位点结合的调控作用的准确预测因子。
值得注意的是,使用ns-sgRNA不会将反式激活降低到与hp-sgRNA相同的程度,这表明发夹的形成而不是简单的sgRNA延伸是调节dCas9结合的原因。但是,平均而言,与未修饰的指南(野生型sgRNA(WT-sgRNA))相比,ns-sgRNA导致基因激活降低了约2.8倍。这与间隔子长度对基于dCas9的转录调节子的效率具有实质性影响的其他证据一致,强调了在测量sgRNA二级结构的影响时需要控制向导长度。实际上,长度效应可能是观察到具有鸟嘌呤二核苷酸延伸的sgRNA具有增加的特异性的根本原因。
这些数据描述了5’sgRNA延伸对SpCas9与DNA结合的非线性影响,这取决于间隔子的长度和二级结构。这种关系通过数据中的三个关键区域来表征(图1c)。
- 首先,对20个核苷酸间隔子的延伸导致整体结合的减少,而这与二级结构无关(图1c,“区域1”)。
- 其次,形成延伸较弱的二级结构的延伸似乎对SpCas9结合的作用似乎不如长度效应所致(图1c,“区域2”)。然而,在该区域仍可能抑制R环的形成。
- 最后,更稳定的发夹会导致hp-sgRNA二级结构的强度随Cas9的结合而下降(图1c,“区域3”)。
- 此外,随着发夹延伸到sgRNA的种子区域( seed region)中,这些活性降低会发生,这对于启动Cas9与靶标之间的相互作用至关重要。发夹结构调节靶基因激活的趋势在人细胞中的另外两个基因靶点上得到了证实(补充图1)。
尽管我们将基因激活的变化归因于hp-sgRNA调节R环形成,但以前的研究通过Northern印迹法显示,有效地将sgRNA的5’延伸加工成20个核苷酸的间隔基[19,33]。为了控制发夹的加工和sgRNA变体的表达,我们重复了该实验,收集了总RNA,并通过定量PCR(RT-qPCR)和5’的逆转录进行了样品匹配的IL1RN和sgRNA表达的测量。通过5’快速扩增cDNA末端(RACE),然后进行RNA测序来处理sgRNA(补充图2a,b)。忠实地复制了IL1RN基因激活中的模式(补充图2c,d)。我们观察到hp-sgRNA表达与hp-sgRNA活性之间没有相关性(补充图2e,f)。
与先前的报道相反,我们观察到hp-sgRNA的加工程度中等至最低,而更强的预测二级结构所经历的加工较少(补充图2g,范围为0.8-48%),与以前的报告相反[19,33]。相应的ns-sgRNA的加工速率更高(补充图2h,加工范围为52-79%)。我们观察到hp-sgRNA加工水平与IL1RN反式激活之间没有明确的关联(补充图2i,j)。这些数据表明,hp-sgRNAs保留在细胞中,可以容纳在Cas9结合袋中,可以防止加工。
2.3 R环形成的动力学模型
hp-sgRNA和ns-sgRNA之间的行为差异表明,间隔子的二级结构是CRISPR活性的关键决定因素。为了更好地了解间隔子二级结构可能如何影响SpCas9行为,我们应用了R环形成的动力学模型并将其通用化,以适应任何种类的错配,任意数量的错配和RNA二级结构(图2a)。链invasion表示为一系列20个离散状态,状态之间的交换概率由3个能量过程控制:
- 基因组靶标(DNA-DNA)的杂交或融化,
- 杂交或融化间隔子与基因组靶标(RNA–DNA)的结合;
- 间隔子二级结构(RNA–RNA)的断裂或形成。
该方法完全根据经验测得的核酸对的热力学值来定义R环形成的动力学(请参见方法)。
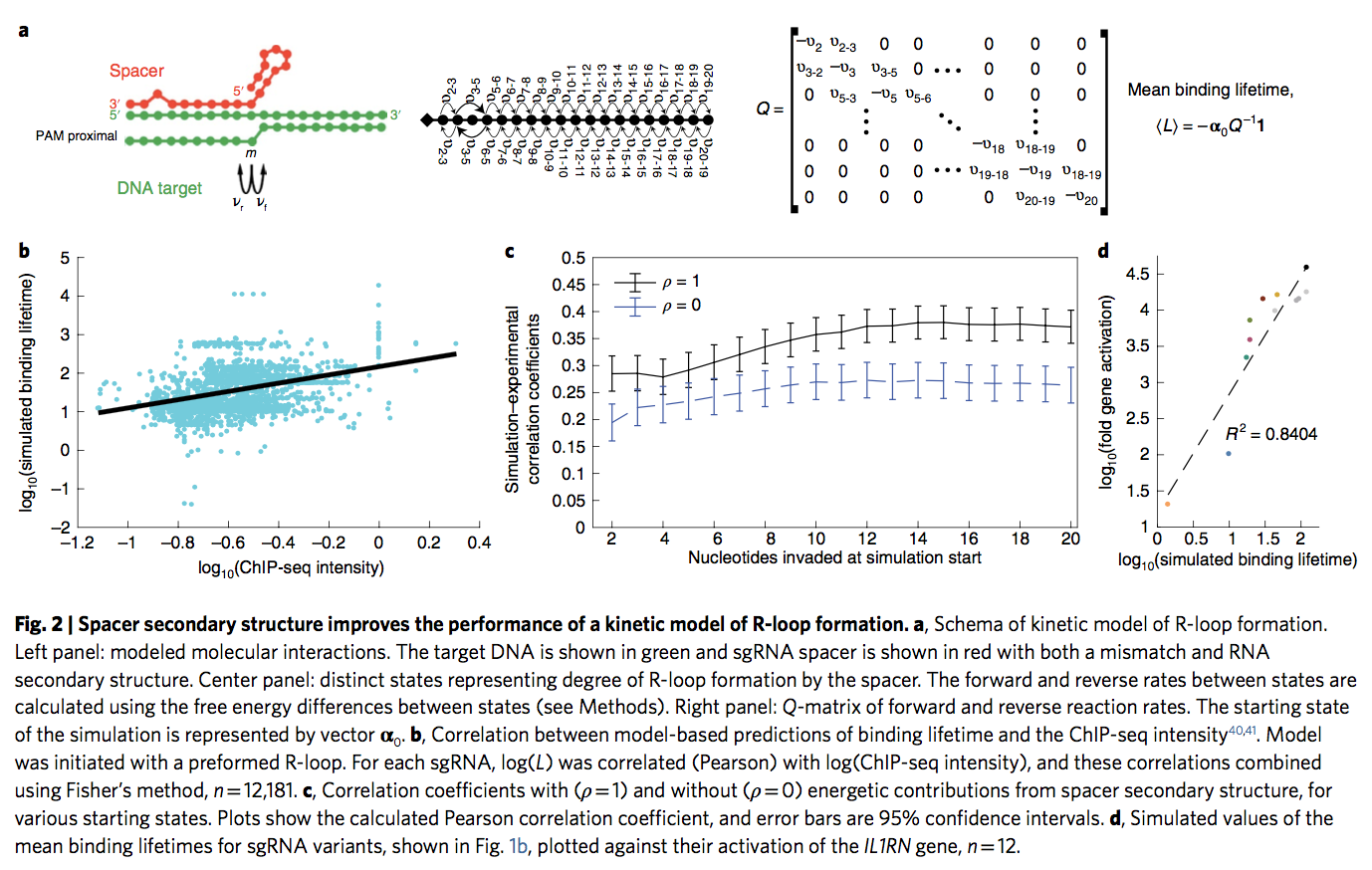
为了测试该模型,我们使用了先前报道的染色质免疫沉淀,然后测序了16个sgRNA和12,181个称为dCas940的结合位点的(ChIP-seq)数据。我们模拟了16种sgRNA各自对每个报告的结合位点的平均停留时间,并将此模拟与测得的ChIP-seq信号进行了比较,并使用Fisher方法将sgRNA之间的相关性进行了比较。当在PAM近端启动模拟时,我们发现相关系数为0.285(95%置信度:0.252,0.317),如果使用预先形成的R环启动,则相关系数为0.380(95%置信度:0.349,0.410)(图9)。 2b)。这些相关性高于以前报道的最佳性能特征,即染色质可及性(chromatin accessibility)。我们模型的预测能力证明R环形成的动力学过程在Cas9与DNA结合中起重要作用。
为了确定间隔子二级结构对模型预测能力的贡献,我们从反应速率中删除了RNA折叠的高能术语。 如果在PAM近端核苷酸处启动模拟,则我们观察到相关性从0.285降低到0.194(95%置信度:0.160,0.228),而如果从 R回路已经形成(图2c)。 最后,我们进行了模拟,以预测用于调节图1中IL1RN启动子表达的hp-sgRNA变体的行为(图2d)。 我们发现估计的结合寿命和基因表达的倍数增加之间有很强的相关性0.915。 总而言之,这些发现表明,间隔子的二级结构通过调节SpCas9核酸水解激活的关键决定因素R环的入侵动力学和稳定性来影响Cas9的结合活性。
2.4 hp-sgRNA增加SpCas9的基因编辑特异性
接下来,我们评估了间隔子二级结构对SpCas9核酸酶活性的影响。我们的假设是发夹结构可以通过调节R环的形成而无需改变与靶位点的结合来增加核酸酶的特异性。因此,对于为SpCas9核酸酶设计的hp-sgRNA,我们通常选择发卡的预测自由能弱于-15kcalmol-1,即在图1c的区域1之内,因为发夹稳定性的任何进一步提高都会导致SpCas9与其目标位点的结合显着降低。为了评估工程改造的hp-sgRNA对人细胞中Cas9核酸酶活性和特异性的影响,我们选择了具有大量特征明确的脱靶位点的间隔子。我们为这些间隔区生成了多种hp-sgRNA,在其中我们改变了几种hp-sgRNA的结构特征,包括利用内部和外部环或调整PAM远端和PAM近侧茎的位置。我们测量了每个间隔物在靶上和靶外位点的插入缺失频率,并将这些hp-sgRNA的活性与未延伸的sgRNA(WT-sgRNA)和截短的sgRNA(tru-sgRNA)的活性进行了比较。我们观察到许多hp-sgRNA设计,它们的靶向活性与WT-sgRNA相当,并且降低了脱靶活性,与tru-sgRNA相当(图3a–c和补充图3–7)。我们通过将靶上突变率除以所有脱靶突变率的总和来定义特异性指标。所有优化的hp-sgRNA均显着提高了SpCas9的特异性,与tru-sgRNA的增加相提并论(图3d和补充图6e)。 EMX1.1间隔蛋白的hp-sgRNA 7具有最高的特异性增加倍数,同时具有间隔蛋白截短和设计的二级结构,表明这些方法在某些情况下可以结合使用(补充图6e)。我们观察到,tru-sgRNA在37个脱靶基因座中的8个处增加了脱靶活性(图3a–c)。这种增加可能是由于tru-sgRNA的序列复杂性降低,并且未发现任何hp-sgRNA变体,与完全抑制方式的hp-sgRNA一致(图3a-c和补充图6a- C)。这些结果共同表明,hp-sgRNA可将SpCas9核酸酶的特异性提高多个数量级。
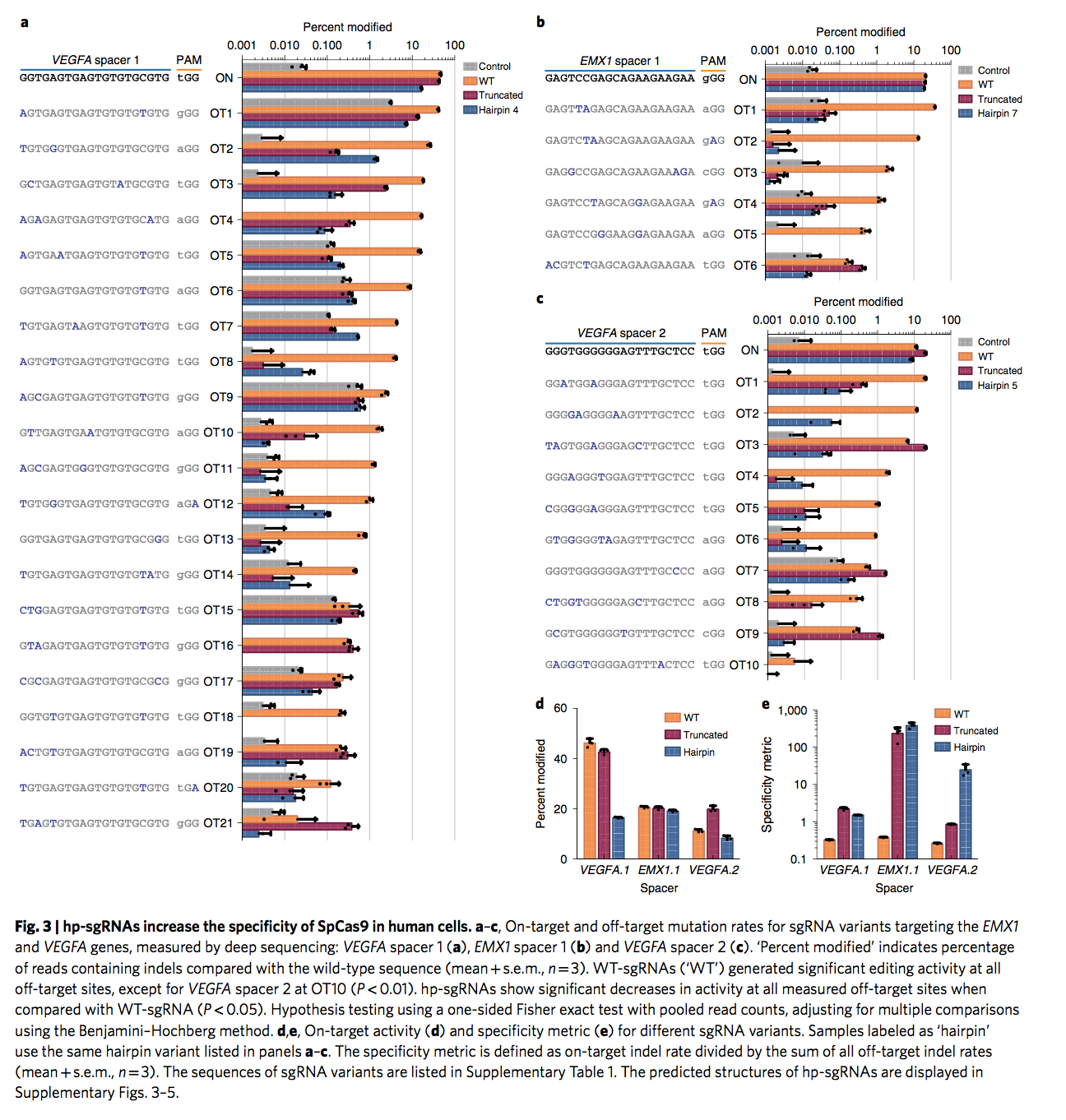
为了测试hp-sgRNA的5’延伸是否可能导致超出先前针对相应WT-sgRNA的鉴定之外的任何新的脱靶裂解事件,我们进行了CIRCLE-seq(环化,通过测序在体外报告裂解效应),一种无偏见的体外方法来确定全基因组裂解事件。我们使用EMX1.1间隔子进行了CIRCLE-seq测序,并使用了WT-,tru-和hp-sgRNA变体。对于每个sgRNA变体,在重复实验中均可靠地鉴定了脱靶位(补充图7a-d)。与WT-sgRNA相比,tru-sgRNA消除了77个脱靶位点,但也有25个独特的脱靶位点,使用CIRCLE-seq可重复检测(补充图8a和9a,b)。相比之下,hp-sgRNA消除了WT-sgRNA发现的124个脱靶位点,并且没有产生独特的脱靶位点(补充图8b和9a,c)。
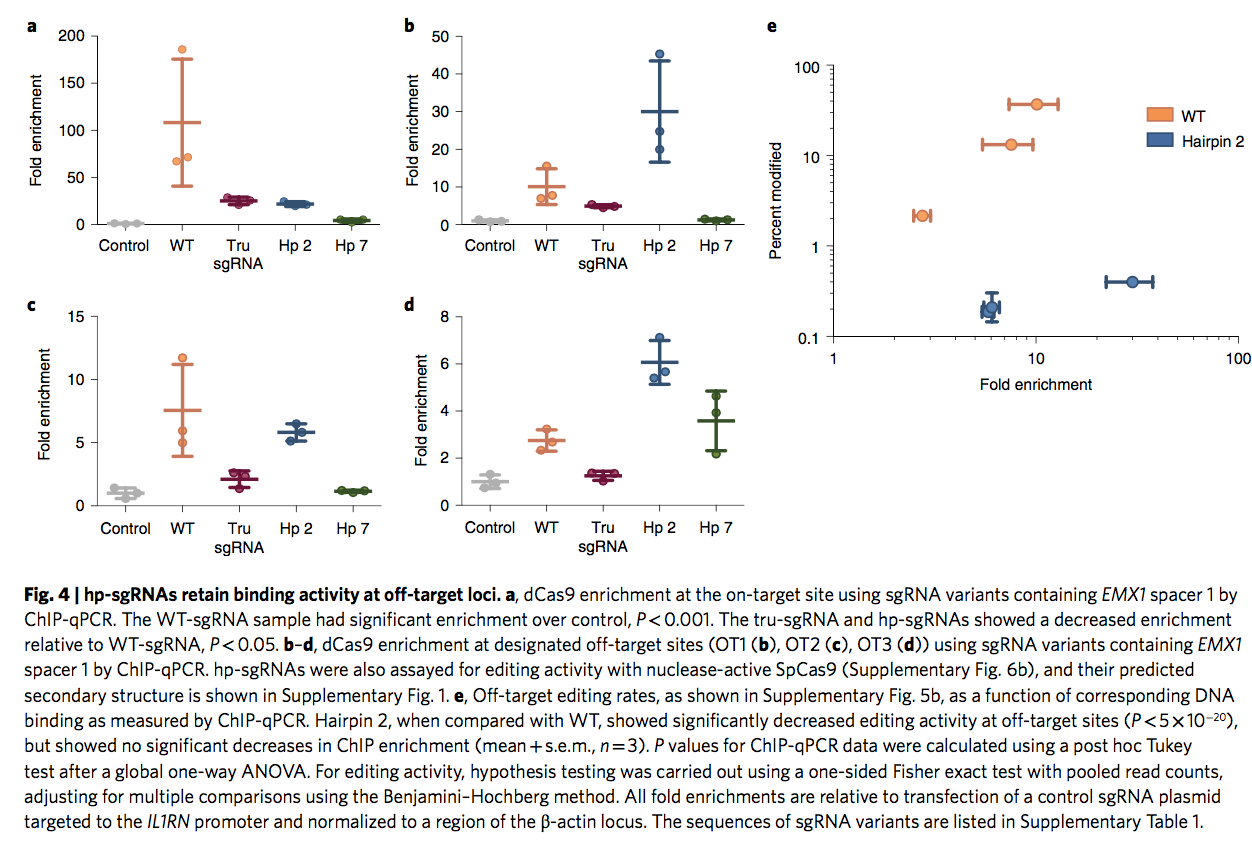
接下来,我们寻求对由hp-sgRNA驱动的特异性增加机制的见解-特别是这是否是与DNA结合力下降的结果。我们用定量PCR(ChIP-qPCR)进行了染色质免疫沉淀,以相同的EMX1间隔子(经核酸酶活性SpCas9测试)测量靶上和靶外位点的核酸酶无效dSpCas9的相对富集。我们观察到,hp-sgRNA和tru-sgRNA在靶点上产生的dCas9占用水平相似(图4a)。有趣的是,即使核酸酶活性降低了一个数量级或一个数量级,相对于WT-sgRNA,hp-sgRNA 2并未显着降低任何测得的脱靶位点的dCas9占有率(图4b–d)。这表明,与高保真Cas9变体24相似,hp-sgRNA不能通过减少结合来提高特异性。 Hp-sgRNA 7具有更多的可变行为,这归因于发夹和截短的间隔子的组合。
2.5 hp-sgRNA增加Cas9和Cas12a变体的特异性
接下来,我们测试了hp-sgRNA设计是否可以扩展到其他CRISPR系统。特别地,我们对SaCas9感兴趣,因为其紧凑的尺寸便于通过AAV载体进行递送,因此对于基因治疗应用非常感兴趣。虽然SaCas9和SpCas9具有许多相似的域和相似的双叶结构,但它们仅共享17%的序列相似性。
着眼于SaCas9和SaCas9-KKH(一种宽松的PAM变体),我们使用具有先前特征性脱靶作用的靶位点设计了不同茎长的hp-sgRNAs。我们将每个SaCas9的sgRNA变体交付给人类细胞,并检测了目标上和目标外位点的核酸酶活性。与SpCas9相似,根据预测的二级结构的强度,hp-sgRNA可调节SaCas9的活性(图5a,b和补充图10a–c)。也使用了不同长度的tru-sgRNA,尽管它们不会消除脱靶活性而不严重影响脱靶活性。较短的截短导致完全消除了脱靶和脱靶核酸酶的活性(图5a,b和补充图10a–c;数据未显示)。
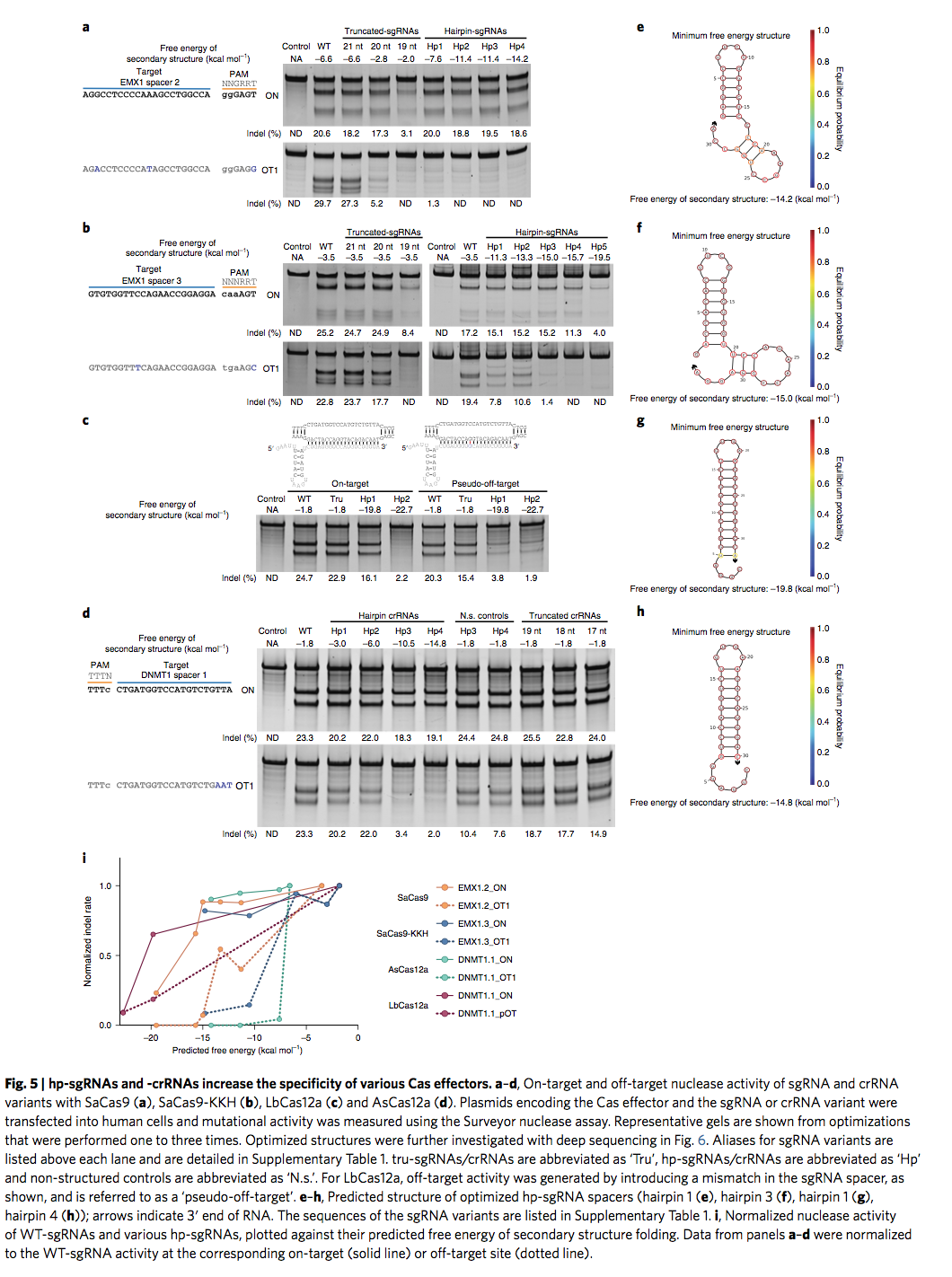
接下来,我们测试了hp-sgRNA是否可用于V型Cas12a核酸酶。尽管SpCas9和Cas12a共享双叶结构,但除了单个RuvC结构域外,它们没有其他结构或序列同源性。 Cas12a核酸酶的独特之处在于它们可以加工自己的crRNA,而这些crRNA足以满足Cas12a靶标的识别和切割。 Cas12a通过位于crRNA 5’末端的发夹识别其crRNA,而间隔子位于3’末端:相对于Cas9 sgRNA结构的反向。与Cas9相比,Cas12a和R环形成机制的目标识别也被逆转:PAM序列位于目标序列的5’端,目标链的R环形成从3’到5 ‘。尽管存在许多差异,我们假设Cas12a核酸酶的活性也可以通过间隔子二级结构来调节。我们使用之前具有脱靶位点的间隔物[14,15,48]设计了具有不同结构稳定性的hp-crRNA。我们观察到AsCas12a和LbCas12a的活性都可以通过间隔子二级结构来调节,脱靶活性可以通过调节二级结构的强度而降低,而不会改变脱靶活性(图5c,d和补充图。 11a–c)。截短的crRNA不能始终导致AsCas12a或LbCas12a的特异性增加,表明该策略可能无法始终如一地转化为Cas12a核酸酶(图5c-d和补充图11a-c)。间隔子的较短截短导致靶标和靶标核酸酶活性的完全消除。我们观察到,hp-crRNAs根据二级结构的强度影响Cas12a核酸酶的活性,这与hp-sgRNAs对SpCas9和SaCas9活性的影响一致(图5c,d和补充图11a–c)。 值得注意的是,随着预测折叠能量的增加,基因编辑活性的降低会优先出现在脱靶基因座处,从而提高特异性(图5i)。
为了确认特异性增加是由RNA二级结构引起的,我们为与Cas9和Cas12a效应子一起使用的hp-sgRNA生成了ns-sgRNA。 对于每个Cas效应子,我们通常选择hp-sgRNA变异体,这些变异体可保持目标活性,但预测的自由能最稳定。 我们提供了这些sgRNA变异体及其各自的Cas核酸酶,并使用深度测序来检测靶标和靶标外位点的突变率(图6a-e)。 在12个间隔序列和6个不同的Cas9或Cas12a变体中,hp-sgRNA的特异性与未修饰的sgRNA相比平均提高了55倍(中位数为12倍),与长度匹配的非结构化sgRNA相比提高了9倍(图 6f和补充图12)。 Hp-sgRNA对具有多个错配的脱靶表现出特别的敏感性(补充图13)。
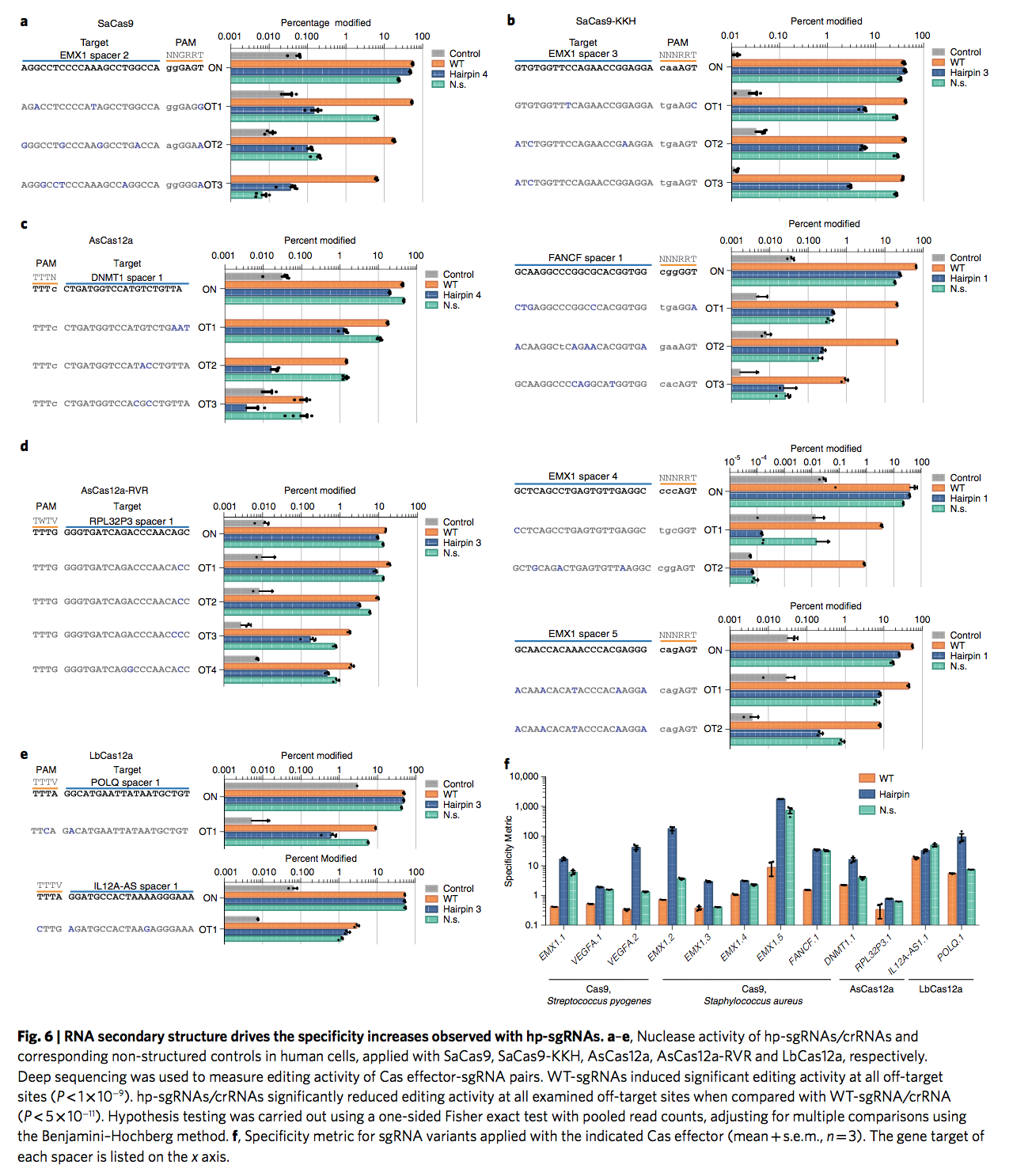
为了进一步确保特异性提高是由于R环形成动力学的调节,而不是转染细胞内表达或稳定性的改变,我们完成了核酸酶活性和DNA结合的体外测定。对于体外核酸酶活性,我们分别通过分别定义浓度的纯化的SpCas9,SaCas9或AsCas12a蛋白与相应的化学合成的WT-,hp-或ns-sgRNA复合,消化了含有目标EMX1间隔物1,EMX1间隔物2或DNMT1间隔物1的PCR扩增子 (补充图14)。在目标位点,相对于SpCas9,SaCas9和AsCas12a的目标位点上的WT-gRNA活性,hp-sgRNA的活性分别降低了85%,59%和69%。相应的ns-sgRNA减少12%,增加35%和6%。 hp-sgRNA的活性在体外而不是在细胞内的靶位上显着降低(图3b,d和6a,c),可能是该检测时间较短或与这些特殊的发夹结构得到优化的细胞内环境。我们还用含有相应脱靶1(OT1)间隔序列的PCR扩增子测试了相同的消化反应。在离靶位点,相对于WT-sgRNA,hp-sgRNA也显示出91%,79%和67%的下降,而ns-sgRNA则下降了88%,38%和0%。为了测定DNA结合,我们使用原子力显微镜(AFM)来直接成像并量化Cas效应子和sgRNA的相同组合在靶上和靶外序列上的相互作用(补充图15)。这些分析表明,相对于目标位点,只有hp-sgRNA而不是ns-sgRNA能够强劲且可重复地降低离靶位点的占有率。总体而言,这些数据支持在体外反应的受控条件下,发夹结构(而不是简单的任何5’延伸)调节CRISPR活性。
三、讨论
CRISPR–Cas核酸内切酶并未进化为可对哺乳动物基因组进行高度特异性的基因编辑,并且迄今为止,已报道了在人类细胞中测试的大多数CRISPR核酸内切酶均存在靶位失活的情况。此外,具有潜在生物技术应用的新型CRISPR系统的发现也在稳定发展。因此,需要改善鲁棒的CRISPR核酸内切酶的性能,并且可以容易地应用于整个CRISPR系统。
如本研究所述,hp-sgRNA的合理设计是满足该需求的一种有前途的方法。对于5个最常用的Cas效应子,利用特征明确的脱靶位点,我们证明了合理设计的RNA二级结构平均可使特异性平均提高55倍。此外,尽管使用的每个Cas效应子具有广泛的生化特性,但我们观察到hp-sgRNA的行为一致,其中CRISPR活性被抑制为二级结构稳定性的函数。
本研究中使用的策略是受以前努力启发的,该努力旨在通过减弱Cas9与DNA之间的直接相互作用来增加核酸酶的特异性。虽然我们没有直接确定hp-sgRNA驱动特异性增加的机制,但我们假设它是通过抑制R环动力学而发生的,R环动力学抑制了CRISPR核酸内切酶在脱靶位点活性所必需的结构转变。证据是三方面的。
- 首先,使用ChIP-qPCR,我们显示hp-sgRNA不会降低脱靶位点的dCas9结合,即使核酸酶活性降低了几个数量级(图4e)。这证明核酸酶活性由于完全R-环形成的抑制而降低。
- 其次,因为RNA-DNA双链体通常被容纳在CRISPR核酸内切酶的中央结合通道中,所以很可能RNA-RNA双链体也被类似地容纳而不干扰RNP复合物的形成。有证据表明具有显着间隔子二级结构的sgRNA可以易与SpCas9配合使用。
- 最后,我们动力学模型的预测能力支持其基本假设:R环的形成是一个由RNA二级结构调节的动力学过程。
总而言之,这些观点表明,sgRNA-核酸内切酶复合物水平得以维持,并且观察到的特异性提高是由二级结构介导的R环形成抑制所致,从而将构象变化限制在脱靶位点处的活化核酸内切酶上。
我们的研究认为R环的形成是控制CRISPR核酸酶活性的主要过程:其调节可进行更特定的基因组编辑,其建模有助于预测CRISPR的活性。对该过程建模的改进将广泛用于计算机模拟脱靶效应和先验设计功能性hp-sgRNA。当我们的模型使用源自体外数据的核酸的热力学参数来近似这种行为时,进一步完善我们对不同CRISPR核酸内切酶催化环境内RNA-DNA相互作用和错配的理解可能会改善其预测和设计性能。最近使用大规模平行评估CRISPR核酸内切酶结合和催化的方法可以为模型改进提供有吸引力的数据集[50,51]。
在这项研究中,我们演示了一种提高跨多种CRISPR系统的特异性的方法。未来的研究将有助于确定hp-sgRNA是否可以类似地调节新的Cas12,Cas13或Cas14效应子[4,5,11,52,53]。调节特异性的hp-sgRNA二级结构可与其他sgRNA工程方法结合使用,以调节活性,特异性和正交性(orthogonality)。 sgRNA工程学,再加上仔细的间隔子选择和优化的基因传递,可以使CRISPR核酸酶具有更高的特异性,可用于下一代基因组编辑,并有助于实现CRISPR在敏感的治疗和诊断应用中的潜力。
四、方法
4.1 质粒和寡核苷酸。
通过Addgene获得用于Cas区段的表达质粒及其各自的sgRNA(Addgene目录号41815、47108、65776、70708、70709、77841、77842、77843、77844); crRNA序列在补充表1中列出,寡核苷酸序列在补充表2中找到。为了创建sgRNA质粒,从IDT获得含有靶序列的寡核苷酸,将其杂交,磷酸化,并使用BbsI或BsmBI位点克隆到适当的质粒中。
所有的hp-sgRNA设计都通过计算机内结构测定获得了信息,并且仅将间隔序列用于这些预测(即,排除了tracrRNA或crRNA中的结构序列)【57】。
4.2 人细胞培养和转染
HEK293T细胞是从美国组织收集中心通过杜克大学癌症中心的设施获得的,并保存在补充有10%FBS和1%青霉素-链霉素的DMEM中,温度为37°C,5%CO2。按照制造商的说明,用Lipofectamine 2000(Invitrogen)转染HEK293T细胞。转染效率通常高于80%,这是在递送对照eGFP表达质粒后通过荧光显微镜确定的。 所有转染均在24孔细胞培养板中进行,并用1:10稀释的聚-1-赖氨酸(P8920 SIGMA)包被。在第1天,包被细胞培养板,每孔接种200,000个细胞。在第2天,将细胞放入Opti-MEM中,并用800 ng质粒(600 ng Cas效应子,200 ng sgRNA)和2μlLipofectamine 2000转染。在第3天,将培养基换成补充有10%FBS和1的DMEM。 %青霉素-链霉素。在第5天收集细胞用于下游分析。
4.3 深度测序
使用DNeasy试剂盒(Qiagen)从细胞中纯化gDNA。对于每种实验条件,从三个单独的转染中产生生物学重复。靶上和靶外位点使用AccuPrime聚合酶(Invitrogen)的100 ng gDNA。引物在补充表3中列出。对于某些区域,PCR中使用4%v / v二甲基亚砜进行有效扩增。 PCR引物包括用于结合Illumina流通池的Nextera衔接子。使用第二轮PCR,特定于组的条形码 添加。使用Agencourt AMPure磁珠(Beckman coulter)纯化所得PCR产物,使用Qubit荧光计(Thermo Fisher)定量,合并并在Illumina MiSeq仪器上与150个碱基对(bp)的配对末端读数进行测序。 CRISPResso用于序列分析【59】。首先修剪序列以去除衔接子序列。使用以下方法过滤序列 最小平均质量得分为30。对读数进行修整以除去衔接子序列。然后使用短读码的快速长度调整(FLASH)合并成对的reads,以创建更高质量的单个序列;使用40 bp的最小重叠。然后使用CRISPRessoPooled对读数进行多路分解并定量非同源末端连接率。最低身分分数为80 用于解复用。由于基于CRISPR的基因编辑很大程度上导致插入缺失而不是取代,因此仅将插入和缺失用于CRISPR产生的非同源末端连接事件。每个生物学复制品每个基因座至少有1,500个reads;平均每个基因座每个重复大约20,000个reads。假设检验是使用单面Fisher精确检验对三个生物重复样本的合并读取计数进行的。使用Benjamini和Hochberg的方法对P值进行多次比较调整。
RT-qPCR, IL1RN激活实验。如所述转染细胞 以上。使用RNeasy Plus RNA分离试剂盒(Qiagen)分离RNA。使用SuperScript VILO cDNA合成试剂盒(Invitrogen)进行互补的DNA合成。使用CBR96实时PCR检测系统(Bio-Rad)使用SYBR green Fastmix(Quanta BioSciences)进行实时PCR,其补充表3中列出了寡核苷酸引物,该引物使用Primer3Plus软件设计并购自IDT。通过琼脂糖凝胶电泳和熔解曲线分析确认了引物特异性。计算适当动态范围内的反应效率,以确保标准曲线的线性。结果表示为通过ΔΔCt方法归一化为GAPDH表达的目的基因的信使RNA表达的倍数增加,由此将对照样品的循环数之差用于归一化实验样品的循环数之差。
…
4.4 Sample-matched 5′ RACE and sgRNA expression measurements
4.5 CIRCLE-seq. CIRCLE-seq libraries were generated largely as previously described
4.6 ChIP-qPCR
4.7 Kinetic R-loop formation simulations
在MATLAB中,通过将过程建模为位置依赖性电位中的一维随机游动,在MATLAB中进行了第一原理的sgRNA入侵DNA双链体的生物物理模拟【29】。 这在MATLAB中被表述为连续时间马尔可夫链。 位置依赖性电位由最近邻依赖性DNA:DNA结合自由能【61】,RNA:DNA结合自由能【62】和引导RNA二级结构自由能决定,随着入侵(invasion)的进行/减少,RNA二级结构自由能被破坏或恢复。 在这里,我们已经概括了该模型,以估计sgRNA在具有任意数量和错配种类的间隔子上的停留时间,并考虑了间隔子二级结构对入侵动力学的影响。
sgRNA与间隔基碱基配对,直至间隔位点m(2≥m≥20)。在每个状态m处,假定sgRNA与DNA处于准平衡状态,因此在完全匹配的间隔位点,正向速率(额外的指导RNA侵入速率; m至m +1)vf使用对称近似法估算为

,其中R是玻尔兹曼常数T是温度(此处为37°C,与我们使用的参数集相对应),其中包括1/2校正项,以满足详细的平衡要求。 ΔG°(m +1)RNA:DNA是RNA与DNA靶点m + 1处碱基配对的自由能。
ΔG°(m + 1)DNA:DNA是间隔区及其互补DNA链之间碱基配对的自由能。
ΔG°(m + 1)RNA,SS是位点m + 1处sgRNA的20-m-1个未入侵核苷酸的预测结构与位点m处sgRNA的20-m个未入侵核苷酸之间的自由能差。
反向速率vr的计算方法类似于

在m = 1时,sgRNA不可逆地从DNA脱落(m = 1充当吸收状态)。使用MATLAB中的rnafold函数计算RNA二级结构自由能。
为了估计存在错配核苷酸时从位点m的转变速率,鉴定出下一个互补位点n,并根据sgRNA(Rm)-DNA靶标(Pm)双链体之间的自由能差异,估算ΔG°(n)MM从位点1到m和sgRNA-DNA靶标双链体从位点1到n。这些双链自由能是使用MATLAB rnafold函数使用序列Rm–UUUU–Pm计算的,最小环的大小(以核苷酸为单位)设置为4。然后将正向速率计算为
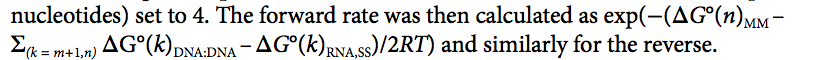
反之亦然。
计算正向和反向速率并将其组装成19×19 Q矩阵(Q),计算sgRNA与间隔子相互作用的平均寿命L为L = –α0Q-11,其中1是19个元素列向量全为1。α0是一个19个元素的行向量,其中包含初始状态的小数填充(m = 2-20)。使用来自Kuscu等人41和Wu等人40的公开数据集对所有16种sgRNA和12,181个ChIP-seq命中物进行了这些实验。对于每个sgRNA,将log(L)与log(相对于目标位点标准化的ChIP-seq计数)相关联(皮尔逊),并使用Fisher方法将这些相关性组合在一起。
4.8 Protein purification.
4.9 In vitro digestion.
参考资料
- Published: 15 April 2019. NAtuRE BIotEChNoloGy. Increasing the specificity of CRISPR systems with engineered RNA secondary structures
