【7.1】通过免疫信息学设计T细胞表位疫苗
1. 摘要
接种疫苗通常被认为是预防传染病最有效的方法。所有疫苗接种都通过向免疫系统呈递外来抗原以引起免疫反应。疫苗的活性剂可以是完整但灭活(“减毒”)形式的致病病原体(细菌或病毒),或已发现具有高度免疫原性的病原体的纯化成分。在分子水平上对抗原识别的更多理解导致了合理设计的肽疫苗的开发。肽疫苗的概念是基于 B 细胞和 T 细胞表位的鉴定和化学合成,这些表位是免疫显性的,可以诱导特异性免疫反应。生物信息学技术和应用的加速增长以及大量实验数据催生了一个新领域,称为免疫信息学。免疫信息学是生物信息学的一个分支免疫学数据和问题的计算机分析和建模。本文综述了不同的基于序列和结构的免疫信息学方法。
2. 介绍
Edward Jenner 在 1796 年首次使用“疫苗接种”一词来描述天花疫苗的注射 [ 1 ]。路易斯巴斯德通过他在微生物学方面的创新工作发展了这一概念。现在,疫苗接种是施用抗原剂以刺激个体的免疫系统并发展对疾病的适应性免疫。疫苗可以改善甚至预防感染的影响。疫苗接种一般被认为是预防传染病的最有效的方法【2 ],和疫苗接种的功效已经被广泛研究和验证[ 3 - 5]。某些疫苗的给药是在患者已经被病原体感染后进行的。据报道,在暴露于天花后的前 3 天内接种疫苗可显着减轻疾病,暴露后长达一周的接种能够提供一定程度的疾病保护,或可能减轻其严重程度 [ 6 ]。此外,最近开发了一种多阶段结核病疫苗,用于在接触病原体后提供保护 [ 7 ]。有许多疫苗的例子,包括针对艾滋病、癌症和阿尔茨海默病的实验性疫苗。所有疫苗接种背后的核心机制是疫苗能够以比病原体本身更快的方式启动免疫反应。
每次接种疫苗的目的是向免疫系统呈递特定抗原或一组抗原,以引起相关的免疫反应。疫苗的主要活性成分可能是无活性的,但仍然完整(减毒的细菌或病毒),或已知会诱导免疫反应的病原体的纯化成分。
3. 疫苗种类
3.1. 灭活疫苗
这种类型的疫苗由在细胞培养物中生长的病毒颗粒组成,并通过应用高温或甲醛等化学物质灭活。病毒颗粒由于被破坏而无法复制,但病毒的衣壳蛋白仍保持完整,足以被免疫系统识别和使用,以诱导反应。如果生产得当,疫苗不会构成威胁;然而,如果灭活不成功,活性感染颗粒可以与疫苗一起施用。通常需要额外的加强注射以确保免疫反应,因为正确生产的疫苗无法在宿主体内繁殖。
3.2. 减毒活疫苗
减毒疫苗含有低毒力的活病毒颗粒。它们保留了缓慢繁殖的能力,因此在第一次接种疫苗后的一段时间内它们仍然是抗原的连续来源,减少了加强注射以保持足够高的抗原水平的需要。这种疫苗是通过使病毒在细胞培养物,在动物或在次优的温度,使选择更小毒性菌株诱变或通过产生,或靶向在毒力[所需的基因缺失8 - 10 ]。
3.3. 亚单位疫苗
亚单位疫苗仅使用最能刺激免疫系统的抗原成分,而不是针对整个微生物。亚单位疫苗含量主要以必需抗原为代表的事实降低了疫苗出现不良反应的机会。亚单位疫苗在不涉及任何病毒颗粒的情况下将抗原引入免疫系统。亚单位疫苗中抗原的数量可以从 1 到 20 或更多。当然,鉴定最有希望刺激免疫系统的抗原通常是一个耗时的过程,并且可能非常困难。与其他疫苗类别相比,亚单位疫苗通常会引起较弱的抗体反应。最成功的亚单位疫苗之一是含有表面抗原 HbsAg 的乙型肝炎疫苗。11 , 12 ]。
3.4. 病毒样颗粒
病毒样颗粒 (VLP) 疫苗仅由参与病毒结构组装的病毒蛋白组成。它们能够在不存在病毒核酸的情况下自组装成类似于它们所源自的颗粒的病毒,这使得它们完全不具有致病性 [ 13 , 14 ]。与亚单位疫苗相比,VLPs 由于其多价和高度重复的结构,通常具有更高的免疫原性。VLP 已经从属于逆转录病毒科、黄病毒科和细小病毒科的多种病毒中产生。针对人乳头瘤病毒和乙型肝炎等病毒的疫苗是基于 VLP 的疫苗,目前已投入临床使用 [ 15]]。此外,基于相同的方法开发了一种针对基孔肯雅病毒的临床前疫苗 [ 16 ]。VLP 通常在多种细胞培养物中产生,例如哺乳动物细胞系、昆虫细胞系以及植物和酵母细胞 [ 17 ]。
3.5. 类毒素疫苗
类毒素疫苗是针对分泌有害代谢物或毒素的细菌的典型解决方案。当不适或疾病的主要原因是细菌毒素时,通常会使用它们。这种类毒素疫苗是通过用福尔马林处理毒素来生产的,从而使它们失活,并且仍然保留它们的结构以供免疫系统进一步识别。类毒素疫苗的例子是针对白喉和破伤风的疫苗。
3.6. DNA疫苗
DNA 疫苗接种是一种非常新的方法,通过施用基因工程 DNA 来诱导对蛋白质抗原的体液和细胞免疫反应。大多数 DNA 疫苗仍处于实验阶段,并且已经在多种病毒、细菌和寄生虫疾病模型以及一些肿瘤模型中进行了测试。DNA疫苗是一种创新方式进行免疫,带来了许多优点超过常规疫苗和给诱导更广泛的各种免疫应答类型[的可能性18 - 25 ]。DNA 疫苗的风险是有限的 [ 22]。几个研究小组证明,癌症疫苗可有效抵抗癌症相关的抗原的特异性免疫的诱导没有负面副作用,如整合质粒DNA到宿主基因组中或病原性抗DNA抗体[诱导23 - 25 ]。
3.7. 肽疫苗
分子水平上抗原识别知识的改进有助于开发合理设计的肽疫苗。肽疫苗背后的总体思路是基于化学方法来合成已识别的 B 细胞和 T 细胞表位,这些表位是免疫显性的,可以诱导特定的免疫反应。靶分子的 B 细胞表位可以与 T 细胞表位缀合以使其具有免疫原性。第一个基于表位的疫苗是由 Jackob等人于 1985 年创建的。[ 26 ]。他们在大肠杆菌中引入了重组 DNA 并表达了针对霍乱的表位。可以为 T 和 B 淋巴细胞构建基于表位的疫苗 [ 27 , 28]。T 细胞表位通常是肽片段,而 B 细胞表位可以是蛋白质、脂质、核酸或碳水化合物 [ 27 – 31 ]。肽由于其相对容易的生产和构建、化学稳定性和无感染潜力而成为理想的候选疫苗。针对多种癌症的多肽疫苗已研制成功,并进入I期和II期临床试验,临床效果满意。肽疫苗接种通常被研究用于改善和预防性免疫治疗 [ 32]]。然而,为了消除障碍,例如需要更好的佐剂和载体或低免疫原性,还有更多需要改进。尽管如此,当前的努力在克服这些限制并为此方法提供改进方面显示出很大希望。
4. T 细胞表位
表位可被抗原的免疫系统部分识别,特别是可被抗体、B 细胞或 T 细胞识别。表位可能属于外源蛋白和自身蛋白,根据它们的结构和与互补位的整合,它们可以分为构象型或线性型 [ 33 ]。T 细胞表位呈递在抗原呈递细胞 (APC) 的表面,在那里它们与主要组织相容性 (MHC) 分子结合以诱导免疫反应 [ 34 ]。MHC I 类分子通常呈递长度为 8 到 11 个氨基酸的肽,而结合 MHC II 类的肽的长度可能为 12 到 25 个氨基酸 [ 35]]。MHC II 类蛋白结合病原体抗原蛋白水解产生的寡肽片段,并将它们呈递到细胞表面以供 CD4 + T 细胞识别。图1)。如果提供足够数量的表位,T 细胞可能会触发病原体特异性的适应性免疫反应。II 类 MHC 在特化细胞类型上表达,包括专职 APC,如 B 细胞、巨噬细胞和树突细胞,而 I 类 MHC 存在于身体的每个有核细胞上 [ 36 ]。
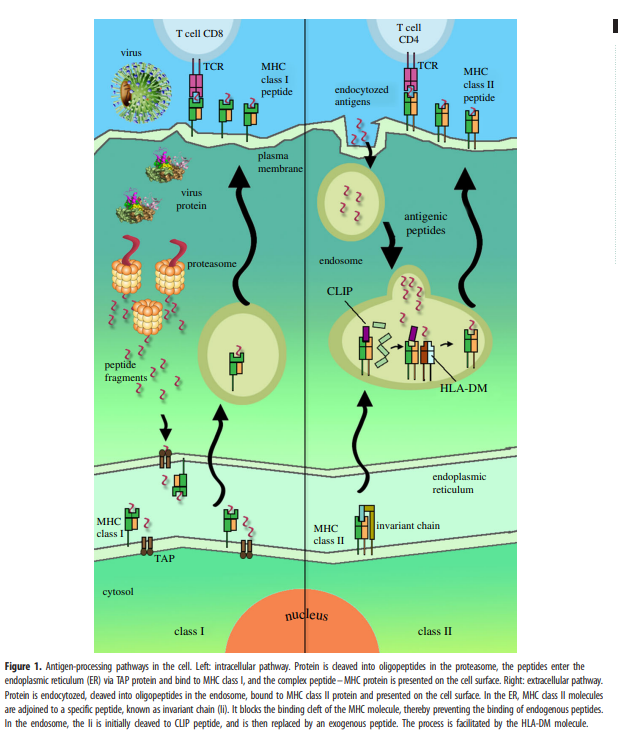
图1。细胞中的抗原加工途径。左:细胞内途径。蛋白质在蛋白酶体中被切割成寡肽,肽通过 TAP 蛋白进入内质网 (ER) 并与 MHC I 类结合,复合肽-MHC 蛋白呈现在细胞表面。右:细胞外途径。蛋白质被内吞,在内体中裂解成寡肽,与 MHC II 类蛋白质结合并呈递到细胞表面。在 ER 中,MHC II 类分子与称为不变链 (Ii) 的特定肽相连。它阻断了 MHC 分子的结合裂缝,从而阻止了内源性肽的结合。在内涵体中,Ii 最初被切割成 CLIP 肽,然后被外源肽取代。该过程由 HLA-DM 分子促进。
T细胞对表位的识别和免疫反应的诱导对于个体的免疫系统具有关键作用。即使是对正常功能最轻微的偏离也会对有机体产生严重影响。在自身免疫性疾病的情况下,T 细胞将细胞的天然肽识别为外来物,并攻击并最终破坏生物体自身的组织。
一些病毒,例如人类免疫缺陷病毒 (HIV)、丙型肝炎以及禽流感和猪流感,依靠各种突变来避免被 T 细胞识别,这些突变有效地改变了病毒基因编码的蛋白质的氨基酸序列 [ 37] , 38 ]。
有关肽表位的知识对于制造基于表位的疫苗具有关键作用,注射到接受者体内后,可以诱导免疫反应。T 细胞表位预测的关键问题之一是 MHC 结合的预测,因为它被认为是 T 细胞识别的先决条件。所有 T 细胞表位都是良好的 MHC 结合剂,但并非所有良好的 MHC 结合剂都是 T 细胞表位。
MHC 是高等脊椎动物中多态性最强的蛋白质之一,在 IMGT/HLA 中列出了 6000 多个 I 类和 II 类 MHC 分子 [ 39 ]。确定这组广泛的等位基因表现出的肽结合偏好超出了目前实验技术的能力,需要开发生物信息学预测方法。迄今为止开发的最成功的 T 细胞表位预测方法是数据驱动的。T 细胞表位预测通常涉及定义特定 I 类或 II 类 MHC 等位基因的肽结合特异性,然后在计算机上预测表位. 使用肽序列数据,实验确定的亲和力数据已用于构建许多 T 细胞表位预测算法。这样的方法包括基于基序的系统,支持向量机(SVM)[ 40,41 ],隐马尔可夫模型(HMM)[ 42 - 44 ],定量构效关系(QSAR)分析[ 45,46 ],和结构-基于方法 [ 47 ]。
5. 免疫信息学
生物信息学技术和应用的加速增长以及大量实验数据对免疫学研究产生了重大影响。这导致了计算免疫学领域的快速发展,并且已经出现了一些以免疫学为重点的资源和软件,它们有助于了解整个免疫系统的特性[ 48 ]。这催生了一个新领域,称为免疫信息学。免疫信息学可以被描述为生物信息学的一个分支,涉及免疫数据和问题的计算机分析和建模。
免疫信息学研究主要侧重于绘制潜在 B 和 T 细胞表位的算法的设计和研究,这加快了实验室分析病原体基因产物所需的时间并降低了成本。使用这些工具和信息,免疫学家可以分析具有潜在结合位点的序列区域,这反过来又会导致新疫苗的开发。分析病原体基因组以识别潜在抗原蛋白的方法被称为“反向疫苗学”[ 49]]。这主要是有益的,因为传统方法需要花时间进行病原体培养和随后的蛋白质提取。尽管病原体生长迅速,但提取其蛋白质然后大规模测试这些蛋白质既昂贵又耗时。免疫信息学能够通过揭示毒力基因和表面相关蛋白来减少相关疫苗开发的时间和资源。
通常,在预测表位时,研究抗原肽与 MHC 分子的结合亲和力是主要目标。实验技术被发现既困难又耗时,因此正在创建并不断改进几种计算机方法来识别表位。方法列表包括矩阵驱动方法、QSAR 分析、结构结合基序的识别、蛋白质线程、同源建模、对接技术以及几种机器学习算法和工具的设计。过去,计算技术只能识别序列特征,但正在设计新的改进算法和工具来提高预测性能 [ 49]。用于开发预测模型的方法可以分为从蛋白质的三维结构中获取信息的基于结构的方法,以及分析氨基酸序列的基于序列的方法。
5.1. 基于序列的方法
5.1.1. 基于主题搜索的方法
在某些肽锚结合位置的优选氨基酸的组合称为基序。该基序搜索是最过时的,但对表位的预测的最广泛使用的方法50 - 53 ]。使用基序文库搜索肽氨基酸序列中的基序[ 54 ]。给定肽的 MHC 结合基序可以通过比较已知的结合剂和非结合剂来确定 [ 55 ]。使用基序搜索方法在恶性疟原虫表达的蛋白质中鉴定结合 HLA-DR 等位基因的表位[ 56]]。EPIPREDICT 是另一种基于基序的工具,用于从参与人类麸质不耐受的蛋白质中鉴定 MHC II 类结合表位 [ 57 ]。达马罗等人。[ 58 ] 开发了计算机程序 MOTIF,它收集了所有已知的 HLA-A*0201 亲和基序。当针对外部测试集进行验证时,该程序识别出 27 种结合剂,随后的实验证实,其中 18 种肽表现出结合亲和力,总体准确度为 61%。另一个工具为E pi中号呃,布朗大学的创建和使用艾滋病毒有关的表位的预测59,60 ]。
一种广泛使用的表位预测工具是 SYFPEITHI,它也是基于模体搜索方法 [ 54 , 61 ]。类似的E pi中号儿的做法,SYFPEITHI被用来评定肽和评价其免疫原性。已经进行了许多体内和体外实验来验证计算机预测 [ 62 – 70 ]。
基于基序的算法的准确度约为 60-70%,主要是因为并非所有的结合肽都具有可识别的基序 [ 71 ]。在许多情况下,预测和实验确定的亲和力之间的相关性非常弱。安徒生等人进行的一项研究。[ 72 ] 将 SYFPEITHI 和 BIMAS 结合剂预测的亲和力与实验确定的一组致癌基因和病毒蛋白的亲和力进行了比较。作者展示了大量错误识别的假阳性,而一些实际表位被预测为非结合物。
5.1.2. 人工神经网络预测
人工神经网络 (ANN) 提供了一种方便的方法来查找关系和描述非线性数据 [ 73 ]。生物信息学研究人员经常使用 ANN 方法来解决哮喘相关问题 [ 74 ]、研究心脏病 [ 75 ] 和药物溶解度 [ 76 ],以及用于表位预测和 MHC 单倍型分析 [ 77]]。申请表位预测时,肽段长度可能变化很大。训练集中包含的序列通常通过分配特定的锚位置来对齐。在构建 MHC I 预测模型时,这是一项微不足道的任务,其中肽长度的差异可以忽略不计,而对于 MHC II 来说,这是一项具有挑战性的任务,其中长度变异性要大得多。
尼尔森等人。[ 78 ] 描述了一种改进的神经网络模型来预测 T 细胞 I 类表位。NETCTL 服务器 [ 79 ] ( http://www.cbs.dtu.dk/services/NetCTL/ ) 使用一种方法来整合肽 MHC I 类结合、蛋白酶体 C 端切割和与抗原加工相关的转运蛋白的预测( TAP) 运输效率。它已从 1.0 版更新到 1.2 版,以提高 MHC I 类肽结合亲和力和蛋白酶体切割预测的准确性。NETMHC 服务器 v. 3.2 [ 80 ] ( http://www.cbs.dtu.dk/services/NetMHC) 基于 ANN 和权重矩阵。它已经接受了来自 55 个 MHC 肽(43 个人类和 12 个非人类)的数据和另外 67 个 HLA 等位基因的位置特异性评分矩阵的训练。MHC I 类分子基序定义明确,但 MHC II 类结合肽的预测被认为更难实现,主要是因为报告的结合肽长度可变、每个肽的核心区域未确定以及一级锚定氨基酸的数量.
5.1.3. 支持向量机预测
SVM 是一组用于数据分析和模式识别的监督学习方法的计算机科学概念,由 Vapnik [ 81 ] 开发,通常用于图像和数据分类和回归分析 [ 82 ]。SVM 属于基于内核的方法组 [ 83]。传统上,SVM 获取一组数据并为每个给定的输入预测它属于什么类型的输入类;因此,SVM 被描述为非概率二元线性分类器。SVM 模型可以表示为空间中的两组点,它们的分布方式是将属于不同类别的两个子集划分为尽可能宽的明显间隙。该模型根据它们落在差距的哪一侧对新数据点进行分类。
SVM 方法的另一个正式描述是它在高维或无限维空间中定义了一个或一组超平面,可用于分类、回归或其他目的。可以通过推导出与属于任何建模类的最近点距离最大的超平面来实现最佳分离。距离越大,模型越可靠[ 84 ]。
Nanni [ 85 ] 展示了使用 SVM 和 SV 数据描述来预测 T 细胞表位。在 TAPPRED 的情况下,Bhasin 和 Raghava [ 86 ] 分析了氨基酸的九个特征,以发现结合亲和力与物理化学特性之间的相关性。他们开发了一种基于 SVM 的方法来预测肽的 TAP 结合亲和力,并发现级联 SVM 更可靠。Cascade SVM 有两层 SVM,其性能优于其他可用算法。实验确定免疫蛋白酶体在 MHC I 类配体的生成中起作用。通常,在研究和预测蛋白酶体的切割特异性时,计算方法优于实验分析。因此,一个名为 PCLEAVAGE [87 ] 已被开发用于预测抗原蛋白中的切割位点。它使用 SVM [ 88 ]、基于并行范例的学习 [ 89 ] 和 Waikato Environment for Knowledge Analysis [ 90 ]。
Sweredoski & Baldi [ 91 ] 提出了 COBEPRO,这是一个用于预测连续 B 细胞表位的两步系统。在第一步中,COBEPRO 使用 SVM 为蛋白质序列片段分配一个片段表位倾向评分。在第二步中,它根据抗原序列中肽片段的 SVM 得分计算每个残基的表位倾向得分。它被合并到 SCARTCH 预测套件中。但是,COBEPRO 无法找到抗原和非抗原之间的区别,为了提高功效,它应该与高通量技术一起使用。
5.1.4. 隐马尔可夫模型
Baum等人在 1960 年代后半期最初描述了 HMM 。[ 92 ]。HMM 在 1970 年代中期首次应用于语音识别 [ 93 , 94 ]。在 1980 年代后半期,HMM 发现了它们在生物序列分析中的应用 [ 95 ],尤其是 DNA 序列。从那时起,它们在生物信息学领域变得无处不在 [ 96 ]。
基于HMM的方法被广泛应用在生物信息学和蛋白质组学用于与螺旋二级结构[蛋白质序列的预测97 ],跨膜区[ 98,99 ]和蛋白质同源性分析[ 100 ]。HMM 还用于序列比对 [ 101 ],以及 Pfam 和 SMART [ 102 ] 的蛋白质家族鉴定。出于基因组学的目的,HMM 用于研究基因剪接 [ 103 ]、系统发育树分析 [ 104 ] 和原毛类动物的基因鉴定 [ 105 ]。
张等人。[ 106 ] 开发了 PREDTAP,用于预测肽与 hTAP 的结合。他们使用了带有 sigmoid 激活函数的三层反向传播网络。输入是二进制字符串,代表九聚体肽。此外,他们使用了二阶 HMM。结果既敏感又特异。Mamitsuka [ 44 ] 衍生出基于 HMM 的高精度模型,用于预测对 HLA-A*0201 和 DR1 蛋白的肽结合亲和力。通过使用 Mamitsuka 的方法,Udaka等人。[ 107 ] 其他 MHC I 类蛋白质的衍生模型。布鲁西奇等人。[ 108] 还使用 HMM 对 HLA-A2 家族成员进行结合亲和力预测。分析只包括与蛋白质直接相互作用的氨基酸。HMM 源自该家族的每个等位基因,并且还与其他等位基因结合的肽被用作训练集。测试集包含与相应等位基因结合的肽。
申巴赫等人。[ 109 ] 比较了 HMM、ANN 和定量矩阵 (QM) 所做的预测。扫描了超过 500 个 HIV-1 和 -2 的氨基酸序列,寻找与 A0201 和 B3501 具有亲和力的肽。ANN 模型对 A0201 等位基因显示出高性能,而 HMM 在预测 B3501 结合体方面更成功。随后的实验表明,基于 QM 和 ANN 的模型成功识别了 26% 的表位。
5.1.5. 通过定量矩阵驱动的方法进行预测
QM 类似于扩展基序,为肽中每个位置的每个氨基酸分配系数 [ 110]。原则上,基于矩阵的表位预测可以分为四个步骤:首先,从给定的蛋白质序列中提取所有可能的肽框。其次,将相应的位置和氨基酸特异性矩阵值分配给给定肽框架的每个残基。接下来,将每个肽的侧链值相加或相乘,得出肽的“分数”。最后,根据肽段得分选择肽段。因此,不是简单地计算锚定残基,基于矩阵的算法考虑了肽序列中每个氨基酸残基的相对重要性,如它们对结合的影响。QM 提供了一个具有易于实施功能的线性模型。使用这种方法的另一个优点是它涵盖了更广泛的具有结合潜力的肽,并为每个肽提供了定量评分。它们的预测精度也相当可观。使用基于 QM 的算法预测 HLA II 类配体的能力首次在 DRB10401 分子中得到证明[111 , 112 ]。这些算法将自然加工的肽和 T 细胞表位排在给定抗原的所有可能肽框架的前 2-4% 中,即使它们只拥有一两个锚定残基。然而,更重要的是,证明了肽评分与结合亲和力之间的相关性 [ 111],因此支持了一个潜在的近似,即给定的残基有助于独立于其相邻氨基酸残基的结合。后来,建立了更多基于 QM 的算法,包括 DRB10101、DRB11501、DRB11101、DRB10701 和 DRB10801 分子的算法。其中一些算法的预测能力通过计算机模拟 M13 肽展示库的筛选进行了验证。使用基于 QM 的算法代替纯化的 HLA II 类分子来富集大的 II 类结合肽库 [ 113 ]。
QMs 也用于预测裂解位点,并在 MAPPP [ 114 ] 中实现。类似的算法应用于预测 B 淋巴细胞的线性表位。Alix [ 115 ] 计算了 20 种常见氨基酸的分子特性(侧链柔韧性、亲水性和可接近的表面),并使用这些特性预测蛋白质中可能与 B 细胞结合的潜在表位区域。
BIMAS 是一种 T 细胞表位预测服务器,它实现了基于 QM 的算法[ 116 ]。BIMAS 用于鉴定各种潜在表位 [ 64 , 70 , 117 , 118 ]。QM 源自 MHC-肽复合物解离半衰期的实验数据。预测与 HLA-A*0201 等位基因结合的模型基于作者的数据,其他等位基因的模型基于文献数据。BIMAS 和 SYFPEITHI 等服务器在预测已知表位方面表现良好,但在筛选蛋白质以寻找未知和新表位时足够准确 [ 69 ]。
另一种基于 QM 的模型是 EpiMatrix,由布朗大学开发 [ 59 ]。它已被用于鉴定 HIV-1 抗原 [ 59 , 119 ]。ClustiMer 和 Conservatrix 中实施了其他类似的方法。ClustiMer 识别混杂(对于给定的 HLA 超家族)肽,而 Conservatrix 确定同一物种突变病原体蛋白质中未改变(保守)的区域 [ 120 ]。
另一类 QM 是位置特异性矩阵,其中计算给定氨基酸出现在某个位置的频率,用于与 MHC 肽结合和不结合 [ 121 ]。尼尔森等人。[ 78 ] 推导出 MHC I 类和 II 类表位的 QM,以解释 Gibbs 能量的变化。
虚拟矩阵 (VM) 是另一种类型的 QM,由 Sturniolo等人创建。[ 122 ]。VM 模拟了每个氨基酸与结合槽口袋之间的相互作用。优势来自于 VM 对具有相似结合槽结构特征的不同等位基因的适用性,而 QM 严格特定于给定的等位基因。TEPITOPE 基于 VM,可预测作为 HLA-DR 结合剂的肽。TEPITOPE 用于鉴定肿瘤抗原 MAGE-3 中的表位 [ 123 , 124 ]。另一个使用 VM 的工具是 ProPred,由 Singh 和 Ragava [ 125 ]创建,其中由 Sturniolo 创建的 MHC 蛋白袋的配置文件作为模型的基础。
MHCPred 是一个基于序列的服务器,使用加法方法 [ 126 ] 来开发 QM。加法方法通过偏最小二乘 (PLS) 方法使用多元线性回归推导出 QM。MHCPred 被用于设计超级结合物 [ 127 ] 并鉴定第一个与 HLA-Cw*0102 结合并源自 HIV 蛋白质组的 T 细胞表位 [ 128 ]。
EpiJen 是一种用于 T 细胞表位预测的多步算法。它模拟了抗原加工的四个步骤 - 在蛋白酶体中切割、与 TAP 蛋白结合、与 MHC 蛋白结合以及被 T 细胞识别 [ 129 ]。对于每一步,都开发了一个 QM,并以连续模式排列,以仅选择那些将由蛋白酶体产生、由 TAP 转运、与 MHC 结合并被 T 细胞识别的肽。在最后一组中收集最有可能充当 T 细胞表位的肽。
VaxiJen 预测全蛋白的免疫原性。它包括通过基于 PLS 的判别分析得出的五个模型,涵盖细菌、病毒、肿瘤、寄生虫和真菌界 [ 130 ]。这些模型显示的准确度在 70% 到 97% 之间。
EpiTOP 是基于蛋白质化学计量学的 MHC II 类结合预测服务器 [ 131 ]。蛋白质化学计量学是一种 QSAR 方法,专门设计用于处理与一组相似蛋白质结合的配体 [ 132 ]。目标蛋白质的结构由适当的描述符描述,并输入 QSAR 的 X 矩阵。肽对特定 MHC 蛋白的亲和力被认为是结合肽和靶蛋白结构的函数。EpiTOP 是 MHC II 类结合预测的前三名最佳工作服务器之一 [ 131 ]。
定量模型的主要缺点是它们强烈依赖于构成肽训练集的数据的类型、数量和质量。包含新数据通常会改变 QM 所基于的值。布鲁西奇等人。[ 133 ] 建议将可靠模型推导的阈值为 150 个肽段作为先决条件,而训练集的理想大小应达到 600 个肽段。然而,实际上,大多数等位基因都由很少超过 50 个肽段的稀缺数据表示。这限制了这种方法对已充分研究的等位基因的适用范围。
5.2. 基于结构的方法
基于结构的方法不仅仅依赖于结合数据和序列信息,而是使用结构信息,并使用结构生物学领域开发的计算方法来预测潜在的良好结合物。
对于识别抗原肽的 MHC 分子,受体和配体之间的几何和静电互补性对于稳定复合物的形成至关重要。许多试图解开控制肽与 MHC 结合的规则的计算研究使用 MHC 结合肽的序列。通过比对已知结合给定 MHC 分子的序列,可以沿着肽鉴定有利于结合的残基。这方面的知识与从晶体学研究一起得到的合成,导致更好地理解的基本原理使得导向肽-MHC识别[ 134,135 ]。
5.2.1. 多肽对接及多肽文库筛选
近年来,许多技术和方法,例如组合肽库筛选和配体对接,常用于药物设计领域,已在生物信息学中得到应用。达文波特等。[ 136 ] 通过评估给定氨基酸对整体肽亲和力的贡献,生成了 MHC II 类模型。他们考虑了氨基酸在某个位置出现的频率。基于源自肽库的关系发现了对 DRB1*0101 表现出亲和力的新肽 [ 137 ]。肽库的筛选也用于研究其他 MHC 等位基因。斯特林等人。[ 138] 通过使用肽库分析了 MHC I 类结合剂的肽特异性。史蒂文斯等人。[ 139 ] 使用肽库来确定鼠 MHC 等位基因的首选肽长度。通过使用组合肽库的位置筛选,Udaka等人。[ 140 , 141 ] 表征与 H-Kb Db 和 Ld 等位基因结合的肽。筛选不同氨基酸在训练集肽的不同位置出现的频率,并生成 QM 以预测测试集肽的亲和力。预测的准确率达到了 80%。Sung等人进行了类似的研究。[142 ] 和 Nino-Vasquez等人。[ 143 ]。
计算机模拟配体对接是一种用于研究分子间相互作用的快速而强大的技术。一般来说,对接模拟的目的有两个:找到给定配体-受体对最可能的平移、旋转和构象并列,并评估配体与其受体的相对结合亲和力。
对接以其在计算机辅助药物设计中的广泛应用而闻名 [ 144 ]。然而,这种方法在设计对 MHC 表现出结合亲和力的新肽方面发现了它的应用。最初,对接研究主要用于研究结合 MHC I 类分子的肽 [ 145 , 146 ]。曾庆红等人。[ 147 ] 通过将具有不同特性(极性、疏水性、带电等)的残基与受体结合槽的不同位置对接,使用具有不同特性的残基,从而评估潜在表位每个位置最可接受的残基特性。另一项研究 [ 148] 使用遗传算法推导出 A2 和 A24 等位基因的 QM,并设计了具有高结合亲和力的肽。肽结构被建模并与结合槽对接。结合能计算为静电和疏水成分的总和。在对肽的结合亲和力进行实验测定后,在预测值和实验导出值之间观察到良好的相关性。
对接也用于研究结合 MHC II 类等位基因的肽,以鉴定锚定位置和暴露于溶剂的位置 [ 149 ]。还通过对接研究了 T 细胞受体和 MHC-配体复合物之间的相互作用 [ 150 , 151 ]。通等人。[ 152 ] 开发一种新的对接方法,包括三个步骤:(i)锚定残基对接;(ii) 肽骨架在结合槽中的定位;(iii) 调整肽主链和侧链的整体定位。与其他方法相比,这种方法显示出更高的准确性。刘等人。[ 67] 在对接模拟过程中考虑到 MHC 蛋白的灵活性。然而,尽管预测精度很高,但这些方法对于在线预测来说并不可行,因为模拟所需的时间太长了。此外,预测的准确性高度依赖于可用于受体的结构信息的质量和正确建模的配体骨架。
EpiDOCK 是一种基于结构的服务器,用于使用基于对接评分的 QM (DS-QM) [ 153 ]来预测肽的 MHC 结合。它预测与 12 个 HLA-DR、6 个 HLA-DQ 和 5 个 HLA-DP 蛋白的结合。
5.2.2. 线程算法的应用
基于知识的线程算法用于区分特定 MHC 分子的结合和非结合肽,而不依赖于以前的数据。该算法通常会考虑沿肽的单个氨基酸的贡献,这些氨基酸促使它们使用基于知识的接触电位来适应 MHC 分子的结合槽 [ 154]]。通常,由于合适的肽主链模板的可用性有限,MHC 结合槽中肽结构的准确预测受到阻碍。尽管如此,通过在肽-MHC 复合物上使用分子建模方法,线程算法的适用性可以扩展到更多的 MHC 等位基因,以预测 T 细胞表位。尽管踩踏无法准确模拟 MHC 凹槽中的肽,但它可以使用结合能评分来验证肽序列在 MHC 凹槽中采用特定折叠的概率 [ 155 – 157 ]。
阿德里安等人。[ 155 ] 研究了 MHC 复合物-肽的相互作用,并揭示了肽的骨架对整体结合剂的选择所起的重要作用。他们还强调了关于配体构象的准确知识的重要性及其对产生更准确预测模型能力的影响。他们使用线程来预测肽的构象,方法是在 MHC 复合物的 X 射线研究中已知的现有骨架上重构它们。用于评估整体结合亲和力的分数是通过将每个氨基酸残基在每个位置的单独结合能分数相加来计算的 [ 158 ]。较低的值对应于较高的亲和力 [ 156 , 157 ]。
这种方法的缺点是,尽管参考肽和测试肽之间存在高度重叠,但一些残基侧链往往指向不同的方向,从而使可预测性变差。然而,额外的建模可能会提高模型的预测准确性 [ 157 ]。
5.2.3. 结合能和分子动力学
可以通过计算配体和受体之间复合物形成过程中游离吉布斯能量的变化来鉴定表位,其定义为游离肽和结合肽的能量之间的差异 [ 159 , 160 ]。可以通过使用评分函数或分子动力学 (MD) 模拟直接比较两种肽的自由能来找到表位 [ 161 ]。MD被用于研究合成肽[结合162 ],MHC肽-蛋白质复合物[ 163,164 ],参与肽-蛋白质复合物的形成[水分子的作用165],A2肽和受体的结合沟[之间的相互作用161,166 ],所述MHC-肽复合物[物的解离167 ],和T细胞受体和肽-MHC蛋白质复合物[之间的相互作用168 ]。罗格南等人。[ 163 ] 模拟了六种肽与 B2705 蛋白的结合,并显示了二级锚定残基的重要性。林等人。[ 169] 通过使用可用的 X 射线结构模拟肽和 HLA-A0201 蛋白之间的相互作用。预测具有高结合亲和力的肽经过实验验证。在另一项研究中,MD 用于确定每个残基在给定位置的贡献,并将结果用于形成用于表位预测的 QM [ 147 ]。进行类似的 MD 模拟以确定 HLA-A*0217 等位基因的锚定残基 [ 170 ]。MD 模拟用于研究与 DRB1 结合的肽 [ 171 ]。戴维斯等人。[ 172] 通过使用模拟退火建立了 MHC II 类蛋白质的表位预测模型,这是一种常见的优化方法,其中肽构象是通过快速升高温度和随后通过在每一步逐渐降低温度来重新计算蛋白质坐标来获得的。所得复合物的能量被导出并用于结合亲和力预测。
另一种方法是将结合能作为溶剂化复合物的能量与溶剂化结合伴侣(肽和蛋白质受体)的能量之间的差值来推导。仅考虑静电和疏水性术语 [ 173 ]。
不同的评分函数可用于评估肽和 MHC 蛋白之间的相互作用。这种方法的优点是它提供了关于哪种类型的相互作用控制复合物稳定性的更准确信息 [ 174 , 175 ]。塞泽曼等人。[ 159 ] 通过使用静电能、溶剂化能和氨基酸侧链的构象熵项,生成描述沿 MHC I 类蛋白结合槽的结合位点的自由能图。弗罗洛夫等人。[ 176 ] 基于极性和非极性相互作用计算八种肽 MHC I 类蛋白质复合物的结合能。夏皮拉等。[ 173 ] 基于三个项——熵、静电和疏水势——计算结合能,并用它来预测小蛋白质复合物的形成。
自由能计算方法也适用于与 HLA-A0201 结合的肽 [ 177 ]。他们使用了一个能量评估函数,其中自由结合能由五个项组成:肽和受体之间的氢键能、疏水原子之间的相互作用能、结合时的熵损失、极性和非相互作用时结合能的降低。 -极性原子,以及原子在具有不同介电常数的环境之间传输所需的跃迁能量。对于另一个实验,Rognan 和同事 [ 174] 使用 Fresno 方法预测自由结合能。训练集包括五个已知的与 HLA-A0201 相互作用的绑定器;有可用于复合物的 X 射线数据和复杂的亲和力数据。基于自由复合能,推导出一个模型来预测另外 26 种结合剂与 HLA-A0204 等位基因的亲和力,该等位基因与 HLA-A0201 具有显着的结构相似性。然而,该研究表明,当有关于受体的结构信息可用时,预测准确性要高得多。该方法用于通过使用可用的 X 射线结构来估计与 A0201 和 B2705 结合的肽的结合能 [ 174]]。随后,弗雷斯诺方法被应用于通过同源性建模构建肽 MHC-蛋白质复合物并计算结合能 [ 175 ]。这种方法的主要缺点是产生结果所需的时间和计算能力,这使得它不适用于在线访问。
六,结论
免疫信息学可以有效地利用计算技术在寻找新疫苗时提供有效和实用的优势。它被认为有助于疫苗设计,因为计算化学有助于药物设计。基于免疫信息学的疫苗设计能够实现疫苗或疫苗成分的有效、具有成本效益的开发。
参考资料
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3603454/ 。2013, T-cell epitope vaccine design by immunoinformatics
