【4.0.1】如果评估预测方法的性能--以基因突变为例
sam点评:
- 数据:(数据量的大小,训练数据和测试数据拆分,benchmarks)
- 評估模型(sensitivity, specificity, positive predictive value, negative predictive value, accuracy and Matthews correlation coefficient; MCC/AUC/FP/FN…)
预测方法越来越多地用于生物科学中以预测不同的特征和特性。二元二态分类器是最常见的应用。它们通常基于机器学习方法。对于最终用户来说,评估计算工具的真实性能和适用性通常是有问题的,因为需要一些有关计算机科学和统计学的知识
一、方法测试方案 Method testing schemes (用于评估的数据集的分类)
三种方法可用于测试方法性能,并可根据可靠性的增加进行分类(图1)。
sam点评:(受限于数据,数据量从小到大,自然验证的的结果也会有差异)

挑战旨在测试是否可以使用现有工具解决某些问题,并找出未来需要什么样的方法。结构预测的关键评估 (CASP) [ 6 ] 是生物科学领域的第一个此类挑战。这个想法过去是,现在仍然是,即使 CASP 已经运行了 20 年,以测试预测方法在不同蛋白质结构相关任务上的表现。方法开发人员在不知道正确结果(盲测)的情况下应用他们的系统,但是挑战评估者可以使用该结果。此设置允许独立测试方法性能。以类似的方式,已经组织了其他关键评估挑战,例如蛋白质功能注释的关键评估(CAFA)[ 7] 和相互作用预测的批判性评估 (CAPRI) [ 8 ]。
CAGI,基因组解释的批判性评估 ( http://genomeinterpretation.org/ ),是基因组变异表型影响领域的方法开发人员面临的挑战。第二个 CAGI 预测季于 2011 年秋季举办。这些挑战的目的不是对预测进行系统分析,而是评估当前可行的方法,提供概念证明,绘制未来工作的指导方向,并确定预测方法可以应用的新领域被需要。
第二种测试策略通常由方法开发人员用来测试他们的方法。这些通常是通过开发人员收集的测试集完成的(尤其是在缺少基准数据集时)并报告某些性能参数。大多数情况下,测试并不全面,并且结果与从其他方法获得的结果无法比较,例如由于使用不同的测试集。有时有选择地提供评估参数,这会导致在确定方法的真正优点和缺陷时出现问题。
第三种方法,系统分析,使用经批准和广泛接受的基准数据集和适当的评估措施来解释方法性能。希望未来变异效应程序开发者能够使用基准测试集和可比较的措施。这已经是多序列比对 (MSA) 领域中的一般做法。
二、分类预测方法 (用于评估的数据进一步的拆分)
大量模式识别方法已应用于生物信息学中的问题,包括基于规则的统计方法和基于机器学习的方法。机器学习的目标是训练计算机系统区分,即根据已知示例对案例进行分类。机器学习方法包括几种截然不同的方法,例如支持向量机、神经网络、贝叶斯分类器、随机森林和决策树。
在下面的讨论中,我们专注于机器学习方法,因为它们现在被广泛用于处理复杂的现象,否则这些现象将难以处理。
- 成功的机器学习方法开发需要高质量的训练集。数据集应代表可能案例的空间。这个空间对于遗传变异来说是巨大的,因为它们可以有如此多不同的影响和潜在机制。
- 另一个方面是机器学习方法的选择。它们之间没有更好的架构。
- 第三,预测器的质量取决于训练是如何进行的,哪些特征用于解释现象和方法的优化。
图2描述了二分类任务中机器学习的基本原理。预测器在一种称为监督学习的方法中使用已知的正面和负面实例进行训练。这导致系统的重组,其细节根据所采用的架构而有所不同。一旦该方法学会了区分案例,它就可以应用于预测未知案例的类别。预测变量可以分类为离散的或概率的,这取决于它们是否为预测提供分数,不一定是 p 值。对于具有离散输出的方法,或多或少是临时的阈值已用于检测最可靠的事件。许多基于机器学习的预测器都是二元分类器,但是,例如通过使用多层两类预测系统,可能有两个以上的输出。
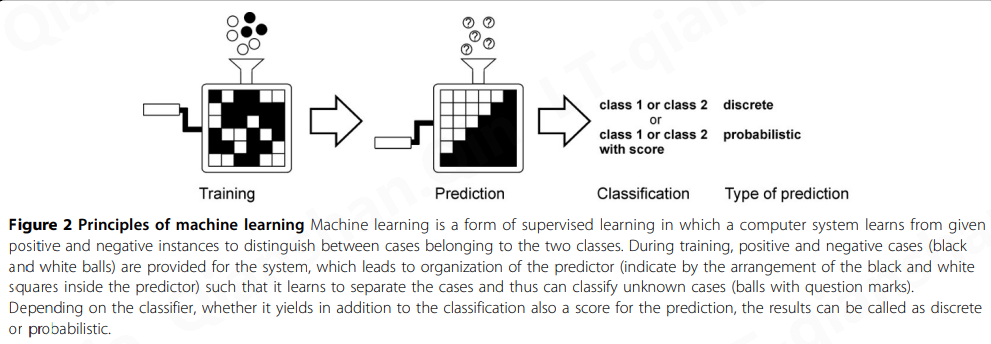
特征描述了所研究现象的特征。如果有几个特征可用,那么选择那些最能捕捉现象的特征就很重要。
- 这部分是由于维数灾难,这意味着当特征数量增加时需要更多的数据。特征空间的体积随着维数呈指数增长,使得数据变得稀疏并且不足以充分描述特征空间中的模式。
- 另一个问题是过度拟合,这意味着学习者由于稀疏数据、复杂模型或过度学习过程而描述了训练数据集中的噪声或随机特征,而不是真实现象。避免过度拟合至关重要,因为它会导致实际情况下的性能下降。
许多类型的生物数据在数量上是有限的。相同的数据不能同时用于方法训练和测试。诀窍是对数据集进行分区。
- 这可以通过不同的方式完成,交叉验证(cross-validation)可能是其中最流行的。数据集被分成k个不相交的分区,其中一个用于测试,另一个用于训练。这是重复k次,直到所有分区都被用作测试集。十个分区即十次交叉验证是最常见的分区方案。从拆分计算的平均性能度量用于描述整体预测性能。
- 随机抽样(Random sampling)是另一种方法,但是,问题是相同的案例可能在测试集中出现不止一次,而其他案例则根本不会出现。
- 另一种计算密集型验证方法是留一法验证( leave one out validation),这是交叉验证的一种极端情况,可对实例总数进行分区。顾名思义,每次只留下一个案例进行验证,而其余案例则用于训练。对于大型数据集,计算要求可能令人望而却步。最后一个方案的一个问题是数据集中是否存在一些非常相似的案例。
通常,训练集在每个类别中应包含大约相等数量的案例。类中案例数量的不平衡可能会在性能评估期间导致问题,如下所述。有一些方法可以处理类不平衡。
三、 方法评价原则
要测试和比较预测变量,必须满足两个要求:
- 必须有已知结果的可用测试数据集
- 并且必须有适当的预测性能评估措施。
Benchmark 是一个黄金标准数据集 - 具有经过实验验证的已知效果的案例,代表了现实世界。这些可用于训练机器学习方法以及测试开发的方法。然而,绝不能将相同的数据用于训练和测试,因为这只会表明该方法记忆示例的能力,而不是其泛化潜力——它在训练集之外的实例上的表现如何。高质量的基准数据集通常需要从不同来源进行细致的数据收集,并仔细检查数据的正确性。
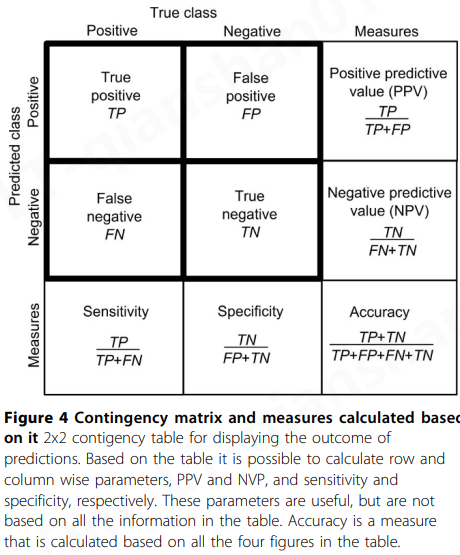
已经开发了许多度量来描述预测器性能,但没有一个度量可以捕获预测器性能的所有方面。将讨论主要使用的措施以及如何解释它们。通常使用预测方法作为分类器来定义案例是否具有调查的特征。这种二进制预测器的结果可以在 2x2 混淆表(也称为列联表或矩阵, contingency table or matrix)中显示。这乍一看似乎很容易解释,但事实恰恰相反,因为必须综合考虑各种复合方面。
基准标准 Benchmark criteria
基准(Benchmark)可以定义为评估的标准或参考,在这种情况下是预测方法的性能。基准测试广泛应用于计算机科学和技术领域。例如,使用标准化基准测试方法测试计算机处理器性能。在生物信息学中,已经有基准,例如 1990 年代的多序列比对方法 [ 9 ]。新型 MSA 构造方法通常使用对齐基准进行测试,例如 BAliBASE [ 10 ] HOMSTRAD [ 11 ]、OxBench 套件 [ 12 ]、PREFAB [ 13 ] 和 SABmark [ 14 ]。其他生物信息学基准包括蛋白质 3D 结构预测 [ 15 – 17]、蛋白质结构与功能预测[ 18 ]、蛋白质-蛋白质对接[ 19 ]和基因表达分析[ 20 , 21 ]基准等。
不同社区之间的基准使用情况各不相同。对于变异效应预测,基准尚未可用,因此作者使用了不同的数据集。随着 VariBench ( http://bioinf.uta.fi/VariBench/ )(Nair和 Vihinen,已提交)的发布,情况最近才有所改变。
一个有用的基准应该满足某些标准。这些标准在不同领域之间有所不同,但也有一些共同特征(图3)。Gray 最初为数据库系统和事务处理系统制定的标准仍然有效 [ 22 ]。MSA 的标准 [ 23] 和变化数据(Nair 和 Vihinen,已提交)基准已经定义。这些包括相关性,这意味着数据必须捕获问题域的特征。可移植性允许测试不同的系统。基准的可扩展性允许测试不同规模的系统,简单意味着基准必须是可理解的,因此是可信的。可访问性意味着基准必须是公开可用的,将任务级别设置在适当级别的可解决性(不太难,不要太难),独立性以保证基准没有使用要测试的工具开发,以及进化及时更新基准。

在考虑变化基准(variation benchmarks)时,数据集应该足够大以涵盖与特定特征或机制相关的变化。
- 例如,在错义变异的情况下,这意味着非常多的实例,因为总共有 150 个单核苷酸变化导致氨基酸替换。
- 要具有统计能力,需要几个案例。随着特征的组合,所需的案例数量呈指数增长。
- 数据集必须是非冗余的,并且没有相似或高度重叠的条目
- 数据集必须同时包含正面(显示调查的特征)和负面(没有效果)案例,以便可以测试方法区分效果的能力。这可能会导致数据收集出现问题,因为某些现象非常罕见,可能只存在少数已知案例。
VariBench 是与变化相关的基准数据集的数据库,可用于开发、优化、比较和评估预测变化影响的计算工具的性能(Nair 和 Vihinen,已提交)。VariBench 数据集提供变异位置到 DNA、RNA 和蛋白质以及 PDB [ 24 ] 中蛋白质结构条目的多级映射(如果可能)。方法开发人员需要将他们的数据集提交给 VariBench,以便分发给社区。
变异本体论(VariO,http ://variationontology.org/ )已被开发用于系统描述和注释变异对 DNA、RNA 和/或蛋白质的影响和后果,包括变异类型、结构、功能、相互作用、特性和其他特征( Vihinen,准备中)。VariO 注释数据将允许轻松收集新的专用基准
四、评价措施 Evaluation measures
二元(致病性/良性)类型预测因子的结果通常呈现在 2x2 列联表(contingency table )中(图4)。正确预测的致病性(非功能性)和中性(功能性)病例数用TP(真阳性)和TN(真阴性)表示,错误预测的致病性和中性病例数用FN(假阴性)和FP表示(误报)。
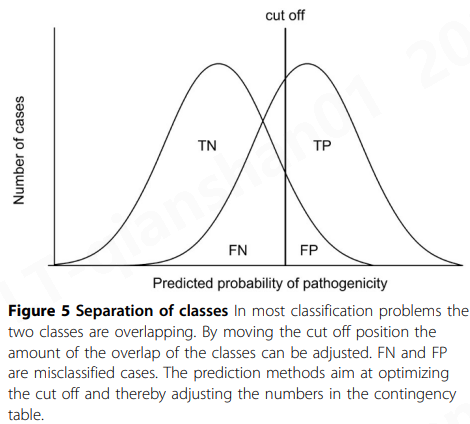
两类预测方法的目标是将正面案例与负面案例分开。因为两个类别的预测通常重叠,所以必须优化区分类别的截止点(图5)。通过移动截断不同数量的错误分类案例, FN和FP出现。通过使用表现良好的代表性数据和训练有素的分类器,可以最大限度地减少错误分类。
基于这四个描述符,可以计算出几个进一步的度量(图4)。灵敏度(Sensitivity),也称为真阳性率 ( TPR ) 或召回率(recall),以及特异性(真阴性率,true negative rate,TNR)显示程序正确识别的致病病例和中性病例的比率。阳性预测值 ( PPV )( Positive predictive value ,也称为精确度)和阴性预测值 (negative predictive value , NPV ) 分别是致病性或中性变异被预测为致病性或中性的条件概率。这些和其他参数的数学基础已被详细讨论 [ 27 ]。
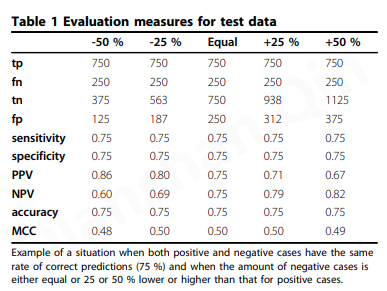
单个参数无法捕获列联矩阵的所有信息。除非使用具有代表性的正例和负例,否则 NPV 和 PPV 的值可能有偏差,甚至毫无意义。通常的要求是数字相等。有时在文献中,数据集非常倾斜。表1表示类不平衡的影响。当阴性和阳性病例总数存在 ± 25 % 或 ± 50 % 差异时,除了均等分布的数据集外,还显示结果用于分析。在用于阳性或阴性病例比率(灵敏度和特异性)的列参数中,不受影响,而 NPV 和 PPV 存在显着差异,它们是基于阳性和阴性病例数的行水平比率个案。在所有示例中,正确预测了 75% 的阳性和阴性病例,因此灵敏度和特异性保持不变。因此很明显,班级规模的不平衡严重影响了 NPV 和 PPV 评估标准。
为了克服类不平衡问题,可以采取不同的方法。一种是将较大类的大小修剪为较小类的大小。也可以在列联表中将正例或负例的值归一化为另一类的总和。在生物信息学中,通常可用的数据量有限,因此人们不愿意删除部分数据集。规范化数据时,请确保现有数据集具有代表性,否则可能会进一步增加集合中的偏差。
精度和 MCC (ccuracy and MCC)
特异性、敏感性、PPV 和 NPV 仅使用列联表中一半的信息计算,因此不能代表性能的所有方面。准确性(图4)和马修斯相关系数(MMC)利用了所有四个数字,因此比行或列方式的措施更平衡、更具代表性和更全面。
MCC 计算如下:
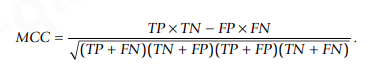
对于此处讨论的所有措施,值越高越好。除MCC外,取值范围为0到1。MCC取值范围为-1到1。-1表示完全负相关,0表示随机分布,1表示完全相关。只有在极端情况下,准确性和 MCC 才会受到类不平衡的影响。
在均匀分布的数据集中,正确预测的案例对参数的影响如图6所示。当 75% 的案例被正确预测时,MCC 的值增长得比其他值慢,达到 0.5。随机结果(50% 的阴性和阳性正确预测)给出的值为 0,而其他参数 - 灵敏度、特异性、PPV、NPV 和准确性为 0.5。图6。可用于检查均匀分布数据集的性能,例如,如果未提供文章中的某些参数。偏差很容易被视为与图中关系的偏差。要获得预测器性能的全貌,重要的是同时评估所有六项指标。
其他参数
其他几个参数可以从列联矩阵中导出。这些没有进一步讨论,因为它们没有在文献中广泛使用,并且可以很容易地从前面提出的六个参数中计算出来。这些包括等于 1-特异性的假阳性率 (FPR) 和等于 1-敏感性的假阴性率 (FNR)。错误发现率 (FDR,False discovery rate ) 为 1-PPV。

其他措施包括例如汉明距离和二次距离( Hamming distance and quadratic distance)(也称为欧几里德距离),它们对于二分类数据是相同的,以及相对熵和互信息(relative entropy and mutual information)
ROC分析
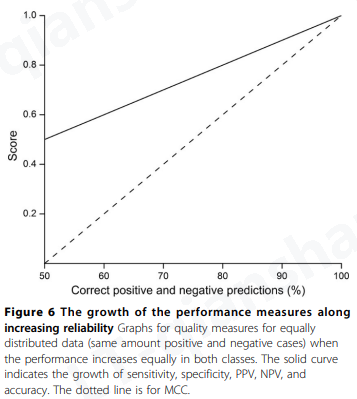
接受者操作特征 (ROC,Receiver operating characteristics) 分析是预测性能的可视化,可用于选择合适的分类器(综述见 [ 28、29 ] )。它表明灵敏度和特异性之间的权衡。当预测变量为概率类型并为分类提供分数时,可以使用特定程序绘制 ROC 曲线。分数通常不是真正的 p 值,而是可用于对预测进行排名的值。
ROC曲线(图7a)是通过首先根据预测分数对数据进行排序来绘制的。然后将数据划分为大小相等的间隔。分区的上限是数据集中的个案数。ROC 曲线在 x 轴 1-specificity ,也称为 FPR,在 y 轴上具有敏感性 (TPR)。
ROC 曲线下面积 (AUC) 已被用作衡量预测优劣的指标(图7a)。它近似于将随机选择的正实例排名高于随机选择的负实例的概率。值为 0.5 表示随机和无用的分类,而 1 表示完美的分类器。请注意,AUC 甚至可以小于 0.5。应该记住,ROC 曲线并不直接表明一种方法的性能。它显示了该方法的排名潜力,这与整体性能有关,进一步强化了这样一个事实,即即使它生成图表,单一度量也无法完全描述预测性能。
如果数据被分为两个以上的类别怎么办?
如果有两个以上的类别,则无法应用上述措施。数据仍然可以呈现在N x N列联表中。一种方法是将数据分成两个类别的多个分区。
如果所有类都需要参数,则有一些可用的选项,但是,单一的度量会有更多问题。可以用与图4相同的方式计算行和列的比率。MCC 实际上是线性相关系数二进制数据的一个特例,它可以以其通用格式用于多个类。在这些情况下也可以使用互信息分析。例如在 [ 27 ]中讨论了适用的措施。
五、性能比较示例
本节讨论变异效应预测方法评估(variation effect prediction )的示例。这些方法包括氨基酸取代(错义变异)耐受性、点变异对蛋白质稳定性的影响以及与 mRNA 剪接相关的变异。讨论集中在比较原则上,特别是根据上述要求的讨论。没有提供实际比较,因为它需要公布报告的大部分内容。由于单个参数不足以用于排名方法,因此读者可以直接阅读原始文章以查找所有详细信息。这里提供了评估参数的方法和使用的总结。
5.1 蛋白质耐受性预测因子 Protein tolerance predictors
单核苷酸改变是最常见的遗传变异类型。人类基因组平均每千碱基都包含这些变异。已经开发了几种计算方法来对这些变化进行分类 [ 1 ]。评估的方法是 MutPred、nsSNPAnalyzer、Panther、PhD-SNP、PolyPhen、PolyPhen2、SIFT、SNAP 和 SNPs&GO [ 5 ]。这些方法在它们考虑的变体的属性以及性质和分类方法上有所不同。Panther、PhD-SNP 和 SIFT 基于进化信息。MutPred、nsSNPAnalyzer、PolyPhen2、SNAP 和 SNP&GO 结合了蛋白质结构和/或功能参数以及序列分析衍生信息。其中大部分基于机器学习方法。
阳性测试数据集包括来自 PhenCode 数据库 [ 30 ]、IDbases [ 31 ] 和 18 个其他 LSDB 的 19,335 个错义变异。阴性数据集由 21,170 个非同义编码 SNP 组成,等位基因频率 > 0.01,染色体样本计数高于 dbSNP 数据库中的 49 个。由于大量的个体预测是致病性或非致病性管道 (PON-P) [ 32 ] 用于将序列和变异提交到分析程序中。
使用上述六项措施评估绩效。这些程序的性能从差 (MCC 0.19) 到相当好 (MCC 0.65) [ 5 ]。
人们普遍认为,有关蛋白质三维结构的信息会提高预测性能。最好的方法也使用结构和功能信息,而其他仅基于序列水平信息的方法表现相当好。
进一步分析成对比较方法,并研究原始或替代氨基酸残基的类型、蛋白质的结构类别或氨基酸替代的结构环境是否对预测性能有影响。
因此,现有程序的性能差异很大,仍然需要更好的方法。考虑到所有评估措施,没有一种方法可以被所有方法评为最佳。
5.2 蛋白质稳定性预测因子
稳定性作为一种基本特性会影响蛋白质的功能、活性和调节。通常发现稳定性的变化与疾病有关。对CUPSAT、Dmutant、FoldX、I-Mutant2.0、两个版本的I-Mutant3.0(序列版和结构版)、MultiMutate、MUpro、SCide、Scpred、SRide等11个稳定性预测指标性能进行了系统的性能评价分析[ 2 ]。预测稳定性中心的 SCide 和 Scpred 以及预测稳定残基的 SRide 仅预测不稳定效应,而所有其他的都评估稳定和不稳定的变化。
蛋白质稳定性信息的主要数据库是 ProTherm [ 33 ]。用于测试的经过修剪的数据集包含来自 80 种蛋白质的 1784 种变异,其中 1154 例呈阳性,其中 931 例不稳定(ΔΔG ≥ 0.5 kcal/mol),222 例稳定(ΔΔG ≤ –0.5 kcal/mol),631 例呈中性(0.5 kcal) /mol ≥ ΔΔG ≥ –0.5 kcal/mol)。大多数方法都是使用 ProTherm 的数据进行训练的,因此只有那些在训练发生后添加到数据库中的案例才用于测试。
在此处推荐的措施中,作者使用了四种,即准确性、特异性、敏感性和 MCC,其余的行参数可以从混淆表中计算出来。
共有三组数据,稳定性增加,中性和稳定性降低。作者通过提供三个结果表解决了多类问题。第一个被分组,使得稳定性增加和降低都被认为是致病的,即阳性。在这些分析中,只考虑了两类,稳定或不稳定和中性案例。
所有案例的结果表明,准确度范围从 0.37 到 0.64,MCC 从 -0.37 到仅 0.12。当单独预测稳定性增加或减少的变化时,所有程序都取得了更好的成功。对于预测稳定性增加和降低变体的最佳方法,MCC 分别达到 0.35 和 0.38 [ 2 ]。
进一步分析了位于不同蛋白质二级结构元件、蛋白质表面或核心以及蛋白质结构类型的变异。
结论是,即使充其量,预测也只是适度准确 (~60%),需要进行重大改进。这些方法的相关性很差。
在另一项研究中,比较了包括 CC/PBSA、EGAD、FoldX、I-Mutant2.0、Rosetta 和 Hunter 在内的六个程序 [ 3 ]。该数据集包含 ProTherm 的 2156 个单一变体。该研究的目的是比较这些方法在 ΔΔG 预测中的性能。因此,他们没有直接预测对蛋白质功能的影响,只是预测自由能变化的程度。唯一使用的衡量标准是实验和预测的 ΔΔG 值之间的相关性。
Dmutant、两个版本的 I-Mutant 2.0、MUpro 和 PoPMuSiC 检测受变异影响的折叠核的能力已得到评估 [ 34 ]。该数据集包含来自 ProTherm 的 1409 个变体,一些方法使用与它们训练过的相同数据进行了测试。他们只使用相关系数作为质量衡量标准。最好在 ~0.5 的范围内。
使用两种蛋白质的数据研究了基于结构的稳定性预测因子 Dmutant、FoldX 和 I-Mutant 2.0 的性能。有 279 种视紫红质和 54 种细菌视紫红质变异 [ 35 ]。视紫红质数据集的最佳预测精度为 <0.60,而细菌视紫红质数据集的预测精度稍高。 铁汉 17:21:10
5.3 剪接位点预测因子
mRNA 成熟是一个复杂的过程,可能会受到许多步骤变化的影响。测试了 GenScan、GeneSplicer、Human Splicing Finder (HSF)、MaxEntScan、NNSplice、SplicePort、SplicePredictor、SpliceView 和 Sroogle 这九个系统的预测行为 [ 4 ]。
测试数据集总共包含 623 个变体。第一个数据集包含 72 个变异,影响 5' 和 3' 剪接位点的四个不变位置。第二个包括 178 个变异,这些变异要么位于非规范位置的剪接位点,要么是远内含子变异,要么是短距离变异。第三组 288 个外显子变异包括 10 个激活隐蔽剪接位点的外显子替换。第四个数据集中是阴性对照,共有 85 个变异对剪接没有影响。
结果仅包含预测案例的数量和正确案例的百分比,因此无法对方法的优点进行详细分析。
作者推荐了一些程序,但指出计算机预测需要在体外进行验证。
六、方法开发人员和用户的清单
提供此清单是为了在比较和测量预测器的性能以及选择合适的预测器时提供帮助。这些是方法开发人员应包含在文章中或作为文章补充的项目,因为它们可以有效地比较和评估预测器的性能。
评估方法性能和比较不同方法的性能时要检查的项目:
- 方法描述的详细吗?
- 开发人员是否使用已建立的数据库和基准进行培训和测试(如果可用)?
- 如果没有,数据集是否可用?
- 是否提到了方法的版本(如果存在多个版本)?
- 列联表( contingency table)可用吗?
- 开发人员是否报告了所有六项性能指标:灵敏度、特异性、阳性预测值、阴性预测值、准确性和马修斯相关系数。如果不是,是否可以根据开发商提供的数据计算?
- 方法测试中是否使用了交叉验证或其他一些划分方法?
- 训练集和测试集是否不相交?
- 结果是否在敏感性和特异性之间取得平衡?
- ROC曲线是基于整个测试集绘制的吗?
- 检查 ROC 曲线和 AUC。
- 在所有措施中,该方法与其他方法相比如何?
- 该方法是否提供预测概率?
参考资料
- 《How to evaluate performance of prediction methods? Measures and their interpretation in variation effect analysis》 https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-13-S4-S2
