【9.1】L1000CDS--LINCS L1000特征方向签名搜索引擎
-
目前,基于网络的集成细胞签名(LINCS,library of integrated network-based cellular signatures )L1000数据集库包含超过一百万种化学干扰的人类细胞系的基因表达谱。
-
通过独特的几种内部和外部基准测试方案,我们证明与当前用于计算L1000签名的MODZ方法相比,使用特征方向(CD,characteristic direction )方法处理L1000数据可显着改善信噪比。
-
CD处理的L1000签名通过称为L1000CDS2的基于Web的最新网络搜索引擎应用程序提供。 L1000CDS2搜索引擎提供了成千上万个小分子签名及其成对组合的优先级,可以使用两种方法预测它们模仿或逆转输入基因表达签名。 L1000CDS2搜索引擎还可以预测通过我们处理的L1000测定法分析的所有小分子的药物靶标。
-
通过计算L1000小分子签名与从基因表达综合(GEO)提取的签名的大量集合之间的余弦相似性来预测目标,以针对哺乳动物细胞中的单基因扰动。我们应用了L1000CDS2来优先处理从GEO提取的670种疾病特征中预计会逆向表达的小分子,并优先考虑可以模拟通过L1000分析分析的22种内源性配体特征的表达的小分子。
-
作为案例研究,为了进一步证明L1000CDS2的实用性,我们在30、60和120分钟的时间收集了感染埃博拉病毒的人细胞的表达特征。用L1000CDS2查询这些特征,我们确定了kenpaullone(一种GSK3B / CDK2抑制剂),我们在随后的实验中显示,它在体外抑制埃博拉病毒感染方面具有剂量依赖性,而不会在人细胞系中引起细胞毒性。总之,L1000CDS2工具可以应用于许多生物和生物医学环境,同时可以改善从LINCS L1000资源中提取知识的能力。
一、前言
系统地收集和分析来自人类细胞系的全基因组基因表达药物反应数据,可用于确定药物再利用的机会,发现化合物的新作用机理,实现内源性配体的小分子模拟,并有助于预测副作用用于临床前化合物。 这种方法最初是通过“连接图”(Connectivity Map)得以实现的,该图包含了四种人类癌细胞系在暴露于〜1300种药物和其他小分子中的一种,6 h后的转录反应数据。通过使用基于Luminex微珠技术(称为L1000)的经济有效的全基因组转录组学测定方法,在基于网络的集成细胞签名(LINCS,LINCS)程序的NIH库的支持下,Connectivity Map项目得到了扩展。 LINCS程序的第一阶段完成后,可获得LINCS-L1000数据,LINCS-L1000数据可提供约50个人类细胞系对各种浓度范围内约20,000种化合物中的一种的响应,总共进行了超过一百万次实验。
从信使RNA表达数据(例如LINCS-L1000数据)中提取签名(signatures)的过程可以使用多种统计方法来完成。当前,LINDS-L1000数据的签名是使用调节Z分数(MODZ, moderated Z-score )方法计算的。最近,我们开发了一种用于计算特征的多变量方法,称为特征方向(CD, Characteristic Direction )。当比较两种情况时,例如,比较药物治疗后的基因表达,CD方法可减轻显示大小变化较大的单个基因的权重细胞与对照细胞。与诸如倍数变化法之类的其他方法相比,某些幅度发生实质性变化的基因可能会获得较低的分数或P值。实际上,倍数变化法仅考虑幅度的变化;并且表现不佳。 CD方法赋予在重复中以相同方向一起移动的基因更多的权重。因此,一个变化不大但“运动”的基因与大量其他基因一起得分可能会比一个整体变化幅度更大的基因得分更高。该方法首先使用线性判别分析来识别能最好地将对照样品与治疗样品分离的线性超平面,然后使用该超平面的法线来定义每个基因表达空间变化的方向。之前我们已经证明,CD方法在识别“正确的”差异表达基因方面,比使用适用于真实数据的几种基准策略的大多数流行的替代方法更为敏感。根据这些基准,CD方法的性能优于limma,DESeq,significant analysis of microarrays 和t检验。
在这项研究中,我们将CD方法应用于处理LINCS-L1000数据。我们证明,与目前可用的计算签名相比,使用CD方法可以显着提取更好的签名。本手稿中提供的所有基准均不同于之前提供的基准。这些新的基准特定于L1000数据,因此,它们通过显示如何使用内部和外部数据评估用于处理L1000数据的计算流水线,为其他方法的开发奠定了基础。为了能够访问和使用经过处理的CD签名的组合,我们开发了基于Web的最新应用程序,该应用程序是一个名为L1000CDS2的签名搜索引擎。 L1000CDS2计算输入特征向量与LINCS-L1000数据之间的夹角,以区分小分子和药物的优先级,以逆转或模拟观察到的基因表达变化。使用L1000CDS2工具,我们优先考虑了可以潜在地逆转从基因表达综合体(GEO)提取的670种疾病特征中的基因表达的小分子,并预测了小分子是内源性配体的潜在模仿者。使用从GEO提取的单基因扰动的基因表达签名的独立收集,我们还预测了通过L1000测定法分析的每个小分子的最可能靶标。
作为案例研究,我们通过实验测试了一个预测。感染埃博拉病毒(EBOV)后不久,来自人树突状细胞的基因表达谱作为输入提交给L1000CDS2。 L1000CDS2签名搜索的最佳候选药物是kenpaullone,这是一种激酶的非特异性抑制剂,包括GS3Kβ和CDK2。随后对EBOV感染的HeLa和人包皮成纤维细胞进行的实验表明,kenpaullone对剂量的抑制作用。使用《京都基因与基因组百科全书》(KEGG)途径进行基因集富集分析,基因本体论生物过程树,小鼠基因组信息学(MGI)哺乳动物表型本体论,ChIP-x富集分析(ChEA),DNA元素(ENCODE),激酶富集分析(KEA)和Expression2Kinases证实了GSK3β和CDK2可能参与了早期EBOV感染,并提示kenpaullone 可诱导先天性免疫应答基因,以协助受感染的细胞检测该病毒。这些结果提示了对抗EBOV和潜在的其他病原体感染的新方法,并证明了L1000CDS2的实用性。
二、结果
2.1 对标特征方向法 Benchmarking the characteristic direction method
正如我们先前所展示的,CD方法显着提高了已发表的微阵列和RNA-seq研究中提取的基因表达签名的质量,我们假设该方法将显着改善L1000数据集中的签名提取。为了检验该假设,我们使用唯一的当前实施的方法(称为中度Z分数MODZ, moderated Z-scores )对用CD方法提取的L1000签名进行了基准测试。首先,我们检查了CD签名P值和MODZ签名强度(distil_ss)的总体分布,该数据来自两个批次(LJP5和LJP6;图1a,b),共进行了8,301个独特的L1000实验。这些直方图显示了用CD方法提取的签名的尾部要胖得多,这表明该方法可以识别出更多潜在的重要签名。实际上,有685个有效签名通过了distil_ss> 6临界值,目前建议将其称为有效MODZ签名,而CD方法调用的有效签名为2,045个,P <0.01。由于两种方法计算出的签名之间存在一一对应的关系,因此我们计算了两种评分方案之间的相关性。我们观察到两种特征评分方法之间存在温和但显着的相关性(图1c)。这表明方法之间存在一些重叠,但也存在显着差异。

如果不考虑剂量,在LJP5和LJP6的8,301次实验中,有1,415种独特的perturbation 条件。我们检查了两种方法计算出的剂量和总体变化之间的总体相关性。该分析的结果表明,与MODZ方法相比,CD方法在剂量和总体变化之间具有更高的相关性(P值= 3.88e-08,t检验;图1d)。当过滤掉所有不显着的扰动时,这种相关性得到改善,并且两种方法之间的性能差异扩大了(P值= 1.1782e-14,t检验;图1e)。对于下一次基准评估,我们分析了来自LJP4批次的数据,其中有四类内源性配体:生长因子;内源性配体。细胞因子干扰素等已被系统地应用于六种乳腺癌细胞系。我们询问了哪种签名提取方法通过扰动类型,时间点和细胞系将签名分开。使用两种无监督的聚类方法:多维缩放和分层聚类( multidimensional scaling and hierarchical clustering ),我们绘制了签名,同时通过其已知的类成员资格为每个签名着色:摄动类型,时间点和单元格(补充图S1和S2)。这些图表明用CD计算的聚类结果更好地反映了有关所应用实验条件的已知生物学知识。上述基准测试方法仅依赖于L1000数据和有关扰动的一些有限知识,这就是为什么我们称这组基准测试为内在的。可以使用外部数据(称为外部基准)来实现其他基准方法。为此,我们接下来询问不同小分子诱导的相似标记是否也具有化学结构相似性
选择该基准的实验是在9个核心细胞系上进行的:HA1E,VCAP,HCC515,PC3,A375,HEPG2,HT29,MCF7,A549,用10μm的μM处理后,在6或24h时测量的基因表达覆盖了20412 小分子化合物。对于相同条件的实验,仅保留通过CD签名P值测得的最强值。为了计算这些实验的基因表达签名之间的相似性,余弦距离用于CD和MODZ签名。欧几里德距离也用作MODZ签名之间相似性的另一种度量。 20,412个小分子的化学结构被编码为166位分子MACCess系统(MACCS, Molecular ACCess System )指纹或扩展连接指纹(ECFP4, Extended-Connectivity Fingerprints)。 Tanimoto评分用于量化指纹相似度,以Tanimoto系数大于0.9作为截止值。尽管用MODZ方法计算的表达特征与微扰剂化学结构的相似性之间存在某种关系,但CD方法能够恢复结构与表达之间更重要的相关性(图2a)。基准测试策略还强调了ECFP4分子指纹方法优于MACCS方法。

下一个外部基准测试方法询问在药物治疗后,已知药物靶标的直接蛋白-蛋白相互作用(PPI)是否也会差异表达。最近的一项利用原始连接图数据集的研究表明,在药物治疗诱导的差异表达基因中通常会发现已知靶标的蛋白质相互作用。CD签名与MODZ签名比较了区分优先表达基因的能力也是药物靶标的直接蛋白质相互作用者。选择LINCS L1000数据中具有至少一个已知蛋白质靶标的756个小分子的特征进行此基准测试策略。通过每种方法CD或MODZ计算的差异表达基因按其绝对差异表达值进行排序,并使用随机游走法与靶标的直接PPI进行比较(图2b)。与使用MODZ计算的签名相比,CD签名的聚集步幅显示出更尖锐的峰,这表明CD从L1000 LINCS数据中检测到已知靶标的更直接的蛋白质-蛋白质相互作用。总之,内在和外在的基准分析清楚地表明,与MODZ方法相比,CD可能是计算签名的更好替代方法。但是,此结果是全球性的。通常,CD方法比MODZ方法在许多数据集上的效果更好,但是在某些特定条件下,MODZ方法可能始终优于CD。此外,在使用CD方法时,例如在为RT-PCR验证选择基因时,考虑单个基因时应格外小心。这是因为CD方法可以对高基因进行评分,这些基因将显示出单变量方法(如学生的t检验)在统计学上不具有统计学意义的变化。尽管CD在区分差异表达的基因和确定应认为重要的特征方面表现更好,但仍需要更多基准。
对于第三个外部基准,我们检查了哪种方法使用从GEO数据库提取的独立的基因表达特征数据集,直接回收已知的药物靶标。我们从哺乳动物细胞中干扰(敲低,敲除或过表达)单个基因的研究中收集了917个独特基因的2206个签名。使用余弦距离,我们对GEO签名与MODZ或CD方法处理的L1000 LINCS数据的相似性进行了排名,并评估了在DrugBank中列出的已知目标的排名。我们看到CD方法对已知目标的排名高于MODZ方法(图2c)。因此,该基准测试还支持CD方法比MODZ性能更好。由于知道小分子的潜在靶标对于许多药物发现应用程序非常有用,因此使用CD方法将所有小分子签名的预测靶标添加为基于Web的L1000CDS2搜索引擎软件应用程序的预测。
2.2 L1000CDS2基于Web的搜索引擎软件应用程序
L1000CDS2 Web应用程序的输入页面包括五个部分:输入文本框;签名示例;配置;元数据和最近的搜索(图3)。 L1000CDS2的入口点是将向上/向下基因列表粘贴到向上/向下基因文本框中,或者将签名粘贴到向上基因文本框中。签名是基因列表,它们的差异表达值用逗号分隔。仅当两个上/下基因集文本框都被填充时,或者当上基因集文本框中都填充了签名时,搜索按钮才会启用。单击搜索按钮将返回结果页面上表格中的前50个签名。示例签名部分包括用户可以作为输入提交的预先计算的签名。 EBOV签名是此寻呼机中描述的基因表达签名。疾病特征包括通过识别对照和疾病样本GSM文件从GEO手动提取的670个疾病特征。配体特征是从LINCS L1000 LJP4子集计算得出的共有配体扰动。通过使用CD方法比较每个细胞系与其余细胞系的基因表达谱,从CCLE基因表达数据计算出癌细胞系百科全书(CCLE)签名。配置部分提供了几个自定义搜索的选项。模拟/反向滑块可在搜索模仿输入签名的小分子或反向输入签名之间进行选择。默认搜索模式为反向。还支持小分子成对组合搜索。用户可以共享其输入签名和元数据,以便其他人可以查询其签名。在元数据部分中,可以输入与输入签名关联的任何元数据。最重要的是,鼓励用户输入至少一个标签以供将来参考。最近的20个搜索存储在“最近搜索”部分中。单击条目将重新加载最近的搜索结果。

在结果页面上,搜索结果呈现为分页表格,每页14个条目(图4)。 每个条目提供有关签名的信息的七列:等级; 得分; perturbation ; 细胞系 ;剂量; 时间点并与输入重叠。 对于基因集搜索,搜索得分是输入DE基因和签名DE基因之间的重叠除以有效输入。 有效输入是输入基因和L1000基因之间交集的长度,因为某些输入列表包含L1000数据集中不存在的基因。 这包括所有〜22,000个L1000基因,而不仅仅是测量的〜1,000个基因。
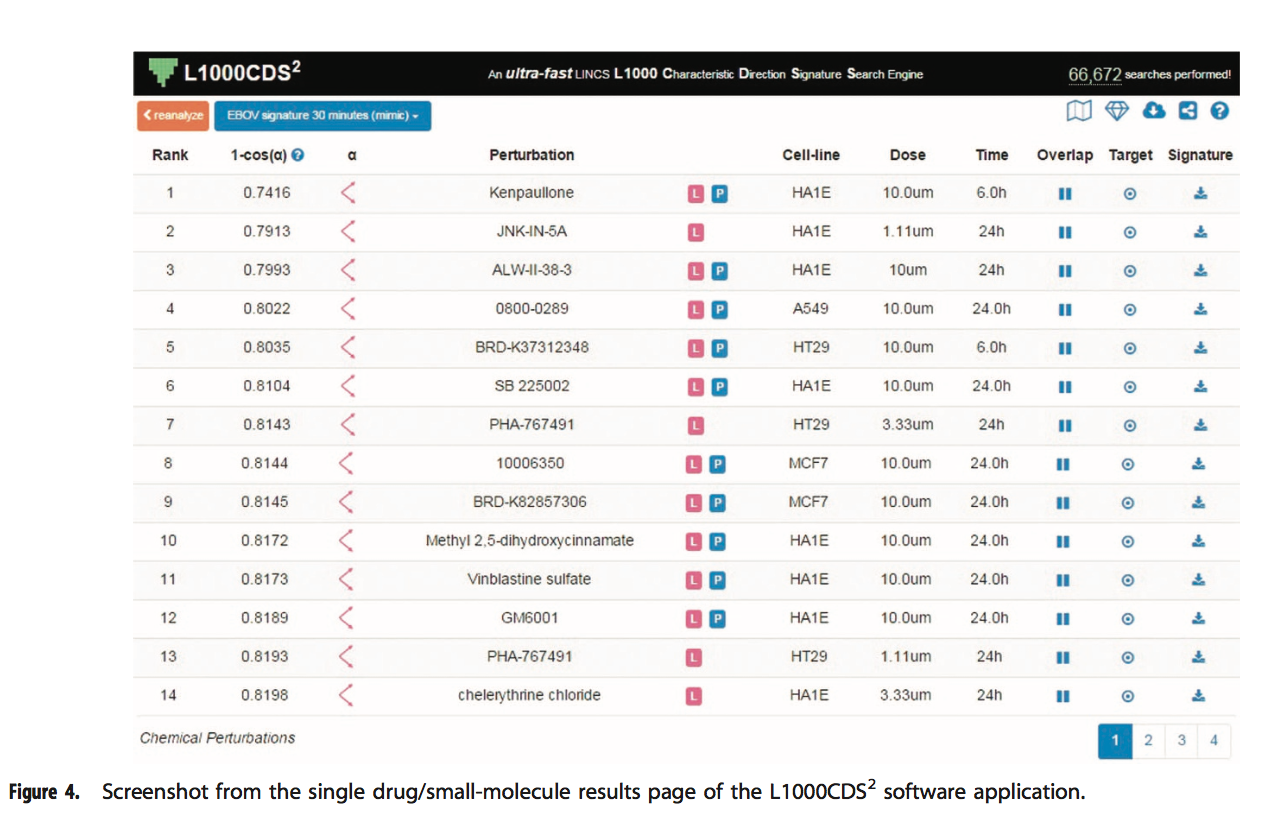
单击重叠按钮将在两个文本框中显示重叠的基因(及其值)。 如果用户输入类型是向上/向下基因列表,则第一个框将显示输入向上基因和签名向上(向下)基因之间的重叠基因,第二个框将显示输入向下和签名之间的重叠基因 在模拟(反向)模式下按下(向上)。 如果输入是签名,则第一个框将显示具有来自输入签名的正值的基因,第二个框将显示负值的基因。 在模拟模式下,两个框中的签名值和输入值都应该是大多数相同的符号,而在反向模式下,则大多数是相反的符号。 每个文本框下方的Enrichr按钮会将基因发送给Enrichr进行富集分析。 单击下载按钮将下载有关签名的所有信息作为JavaScript对象表示法(JSON)文件。
表格顶部是提供各种有用服务的按钮和图标。重新分析按钮将用户重定向到输入页面,其中已提交的列表或签名已预先加载在输入文本框中。然后,用户可以使用不同的配置重新分析其输入,或修改关联的元数据。此功能还影响与他人共享结果。它为用户提供了一种使用不同设置重新分析其输入并为每次分析获取永久URL的方法。标签按钮显示标签和搜索模式。单击按钮显示输入的元数据。云下载图标将结果表下载为.csv文件。菱形图标对排名靠前的小分子的子结构进行富集分析。子结构富集分析的结果显示为表格,其中每一行都是一个显着富集的子结构。每行提供三段信息:子结构,P值和扰动计数。子结构以SMARTS格式表示为字符串。使用费舍尔精确检验计算出P值。扰动计数显示具有相同子结构的扰动数。单击共享图标将生成一个永久URL,可用于通过电子邮件,出版物或其他文档共享子结构丰富化分析结果。单击加号将显示子结构的可视化以及包含子结构的顶部扰动表。该表中的排名与前50个结果表中的扰动排名相同。
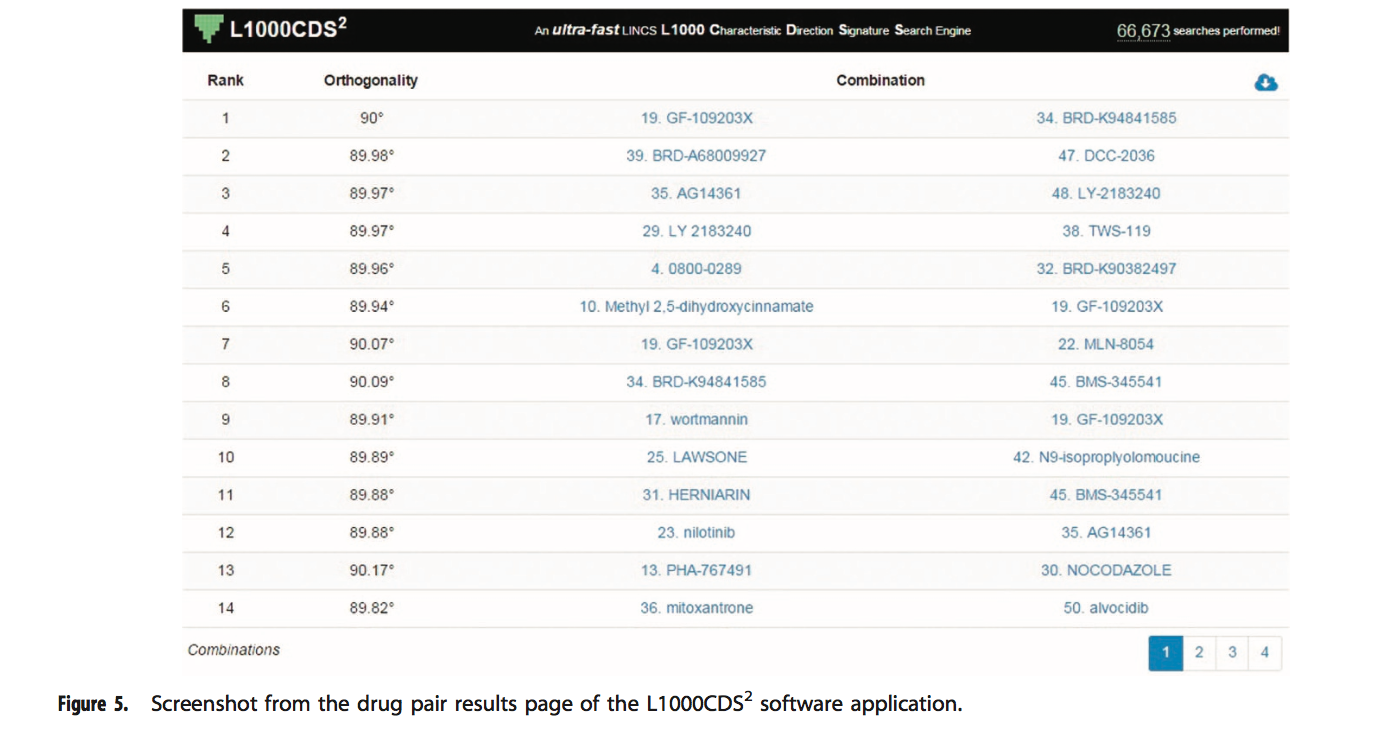
如果用户选择搜索小分子组合,则签名组合表将出现在单个扰动结果表下方(图5)。该表也是分页表,每页14个条目。每个条目提供有关已识别组合的三段信息:等级;协同作用得分和组合。搜索组合时,L1000CDS2在前50个匹配的签名中比较每个可能的对,并通过检查正交性水平来计算每对之间的潜在协同作用。通过基因集搜索,将两个药物特征码的差异表达基因与输入基因列表的重叠重叠计算为协同得分。在余弦距离搜索中,协同作用被计算为两个CD签名之间的正交性。这样做的合理性是,如果两个扰动是正交的,那么它们可能会通过两个独立的途径发挥整体作用。等级基于正交性分数。组合列中每个化学扰动之前的数字是单个签名结果表中该扰动的等级。单击扰动将在单个签名结果表中突出显示该扰动,以便用户可以了解有关该扰动的更多详细信息。单击右上角的云下载按钮将组合表下载为.csv文件。
2.3 L1000CDS2预测kenpaullone是抑制埃博拉病毒感染的潜在药物
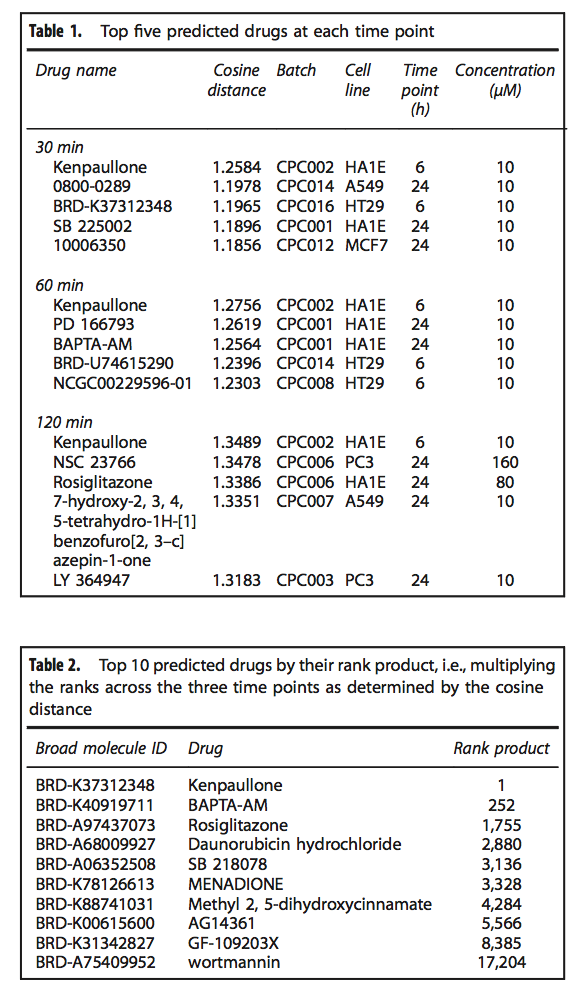
为了进一步利用L1000CDS2的功能,我们将其用于预测可以潜在抑制埃博拉病毒感染的药物和小分子。为此,我们首先从感染了埃博拉病毒的人类细胞中收集了表达特征。用埃博拉病毒感染了来自四个人类供体的树突状细胞,然后在感染前以及感染后30、60和120min对全基因组基因表达进行了测量。计算每个时间点的差异表达基因,并将每个时间点与对照进行比较。这导致1,031个在30分钟时显着差异表达的基因,60分钟时746个和120分钟时的248个差异表达基因。与以前在人类和非人类灵长类动物细胞中感染埃博拉之前和之后其他大多数可用的全基因组表达数据集相比,这些特征为进一步研究埃博拉感染的早期反应提供了机会。我们首先分析了差异使用Enrichr在线工具进行富集分析,表达基因清单。我们发现上调的差异表达基因在免疫应答方面很丰富,并假设模仿该基因表达状态的小分子可能增强内在的免疫应答,以帮助减轻埃博拉病毒感染。我们使用L1000CDS2工具查询了这些签名,以对可以潜在模仿埃博拉病毒感染细胞中基因表达的药物和小分子进行优先级排序。从三个时间点计算每个L1000方向的余弦距离,并带有埃博拉签名方向。然后,按照余弦距离对摄动进行排序,对相似特征进行优先排序。有趣的是,即使每个时间点都有一组独特的差异表达基因签名,肯帕隆在所有三个时间点都排名第一(表1和2)。应当注意的是,使用新的lincscloud Connectivity Map网站上提供的现有签名搜索工具,kenpaullone在60个和120个时间点签名中排名前50位,分别排名第39位和第23位。这些等级相对较高,但可能不足以将肯保隆作为实验验证的首选。
接下来,我们试图评估排名靠前的几个小分子(包括kenpaullone)是否可以抑制组织培养中的埃博拉感染。为了进行初步筛选,我们对HeLa细胞进行了预处理(每个排名靠前的小分子为20μm),然后以5的感染复数(MOI)用埃博拉病毒感染细胞48h。对埃博拉病毒感染的细胞进行病毒抗原染色,并在Opera共聚焦高内涵成像平台上进行分析。在这些条件下,观察到kenapaullone可抑制埃博拉病毒感染约52%(图6a)。 Chk1抑制剂小分子SB218078可抑制埃博拉病毒60%。然而,由于溶解度的问题,使用这种小分子进行的后续研究未得出结论(数据未显示)。其余的小分子AG-14361,甲萘醌和2,5-二氧肉桂酸甲酯对病毒的抑制作用最小,在这些条件下,而柔红霉素则具有细胞毒性。总之,在最初筛选的五个小分子中,kenapaullone仍然是最有前途的。

为了确定kenpaullone是否可以剂量依赖的方式抑制埃博拉感染,我们进行了一系列剂量响应实验。 Hela细胞用肯帕隆(Kenpaullone)预处理,剂量范围为0.3至75μM,然后以5的MOI感染埃博拉病毒48 h。在这些实验中,观察到肯保隆以剂量依赖性方式持续抑制埃博拉感染(图6b,c)。重要的是要注意,尽管该化合物有效抑制病毒感染,但它不影响总细胞数,因此无细胞毒性。由于kenpaullone是已知的GSK3B抑制剂,我们接下来通过实验测试了其他公认的GSK3抑制剂抑制HeLa细胞中埃博拉病毒感染的功效。这些化合物是SB216763,SB415286,TCS2002和TC-G24。如前所述,用药物预处理细胞2小时,然后以5的MOI感染埃博拉病毒。从细胞数量上看,这些化合物是无毒的(只有SB216763在100μm处显示出最小的毒性),但是这些化合物对阻止或增强埃博拉感染的影响却很小。这些结果表明,kenpaullone可能不仅通过抑制GSK3B发挥作用,而且还可能通过CDK2和其他机制发挥作用。我们还通过实验测试了几个小分子,这些小分子被一致预测在所有三个时间点都会逆转表达。这些化合物包括:PAC-1,厚朴酚和NCGC00184902-01。其中NCGC00184902-01显示埃博拉感染略有增强,而其他两种化合物则无效果(图6d)。最后,我们通过测试其对减轻另一种细胞类型的埃博拉病毒感染的功效,来测试肯帕洛酮的作用是否为细胞类型特异性。将人包皮成纤维细胞(被认为是人类的主要细胞系)进行了kenpaullone预处理,并以与HeLa细胞完全相同的方式感染了埃博拉病毒。我们还观察到人包皮成纤维细胞感染的剂量依赖性抑制作用(数据未显示)。
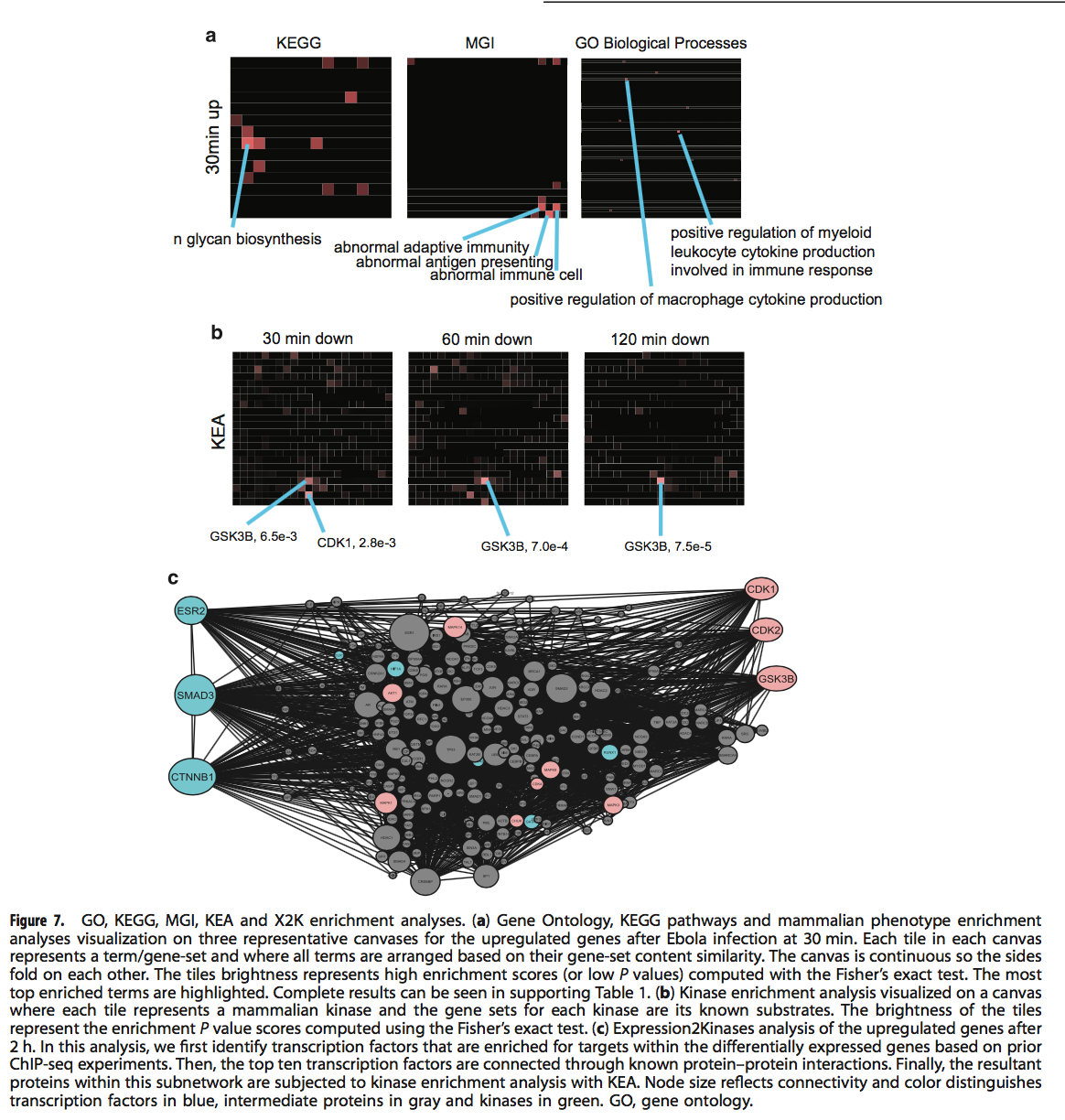
为了确定由埃博拉病毒感染引起的和肯帕洛酮缓解的潜在分子机制,我们进行了基因集富集以及Expression2Kinases(X2K)分析。我们将埃博拉病毒在30、60和120 min时上下表达差异表达的基因作为输入,并使用与kenpaullone匹配的L1000签名。这些基因集已提交给Enrichr22和Expression2Kinases工具进行富集和网络分析。使用基因本体论生物学过程,KEGG途径和来自基因组学的敲除小鼠哺乳动物表型本体论,将第一组富集分析应用于埃博拉病毒诱导的上调基因。小鼠基因组信息学(MGI)基因集库(图7a,补充表S1)。我们观察到一个共同的主题。在所有三个时间点上调的基因与涉及免疫反应和N-连锁糖基化的术语和途径有关。例如,对于60分钟后埃博拉病毒感染后上调的基因,异常的先天免疫是最丰富的MGI小鼠表型术语(校正后的P值<0.0001,Fisher精确检验);而最丰富的KEGG途径是N-聚糖生物合成(调整后的P值<0.00001,Fisher精确检验)。接下来,我们对埃博拉病毒感染后下调的基因进行了KEA13检测。 KEA包括从出版物中提取的激酶-底物相互作用,并从六个激酶-底物开放的在线数据库进行合并。在该基因组文库中,每个术语都是激酶,其底物是与每个术语相关的基因集。有趣的是,在所有三个时间点将KEA应用于下调的基因显示出对已知GSK3B底物(肯帕隆的已知靶标)的富集最多(图7b)。在30分钟的时间点,CDK1和CDK2的底物也与GSK3B一起高度富集。这些观察结果与我们对Kenpaullone作为顶级模仿药物的预测一致。 X2K分析还证实了GSK3B,CDK1和CDK2通过转录因子β-catenin,Smad3和雌激素受体2的潜在参与(图7c)。最近显示肯帕洛酮诱导Smad3活性以促进iTreg分化。23X2K分析首先确定差异表达基因上游的富集转录因子,然后使用从20个数据库中收集的已知PPI连接这些转录因子,然后X2K将KEA应用到连接的转录因子网络。因此,X2K试图通过转录因子和蛋白质相互作用将表达的变化与其上游调节机制和细胞信号通路联系起来。 X2K分析的结果对于不同的阈值和富集测试均具有一致性和鲁棒性。最后,我们询问了kenpaullone和早期埃博拉感染诱导或抑制的重叠基因(补充表S2)。埃博拉病毒感染和肯帕龙均上调的最丰富的基因本体论术语,MGI表型和KEGG途径与先天免疫应答相关,包括以下基因:CIITA,LILRB1,RIPK1,CARD9,ALOX5AP,AHR,TLR4和CTSC。据报道,在两个独立发表的来自ENCODE12和ChEA的ChIP-seq实验基础上,这些基因受STAT3调控。11STAT3是先天免疫应答的已知诱导剂24,我们预测kenpaullone会进一步诱导STAT3活性。但是,这种预测的验证仍有待进一步研究。
四、讨论
总体而言,我们获得了有关早期埃博拉感染可能需要的潜在细胞内途径的初步连贯机制的见解,并确定了可能干扰这一过程的小分子。我们的发现GSK3B抑制剂可以潜在地减轻埃博拉感染,这一点得到了文献中其他证据的支持。先前有报道称,GSK3B抑制剂可降低败血症后的急性炎症反应,保护多器官损伤并减少败血症过程中的肌肉蛋白质降解。已知我们的富集分析所涉及的糖酵解途径与GSK3B活性有关,并且这种增加人们认识到糖酵解途径对埃博拉生命周期至关重要。肯帕洛酮的非特异性使得对其分子机制和直接靶标的研究变得困难。但是,在这里我们观察到肯保隆诱导免疫反应基因的表达,因此它可能是一般的抗病毒候选药物。一项相对较旧的研究表明,干扰素治疗可以改善感染埃博拉病毒的非人类灵长类动物的预后和存活率。然而,已知干扰素会引起严重的副作用,因此可能不大可能产生和释放,这就是为什么小分子例如肯帕龙,其可能潜在地模拟/诱导干扰素。 kenpaullone可以增强先天免疫应答基因的表达,从而有助于感染的细胞和感染细胞附近的细胞更好地感知病毒感染并更快地做出反应。肯帕洛酮是否会干扰病毒的生命周期或只是开启免疫反应仍是一个悬而未决的问题。尚不清楚肯帕龙对病毒先天免疫应答的诱导是通过埃博拉病毒诱导的相同途径还是通过不同的机制。已经开发了更详细的肯帕龙小分子衍生物,29并可以在未来的研究中测试其抑制埃博拉和其他类似病原体的潜能。
总而言之,在这里,我们介绍了一种改进的计算方法,该方法潜在地提高了新生成的公共可用 LINCS-L1000 数据集的子集的实用性,从而可以快速确定可能会逆转或模拟疾病和其他生物学环境中表达的小分子的优先级。通过CD方法计算出的大量签名是作为已经广泛使用的基于Web的最新应用程序提供的。除了优先考虑小分子以逆转或模仿输入特征或针对670种疾病的预先计算特征以及内源性配体的集合外,基于L1000CDS2网络搜索引擎的工具还可以预测成对的小分子组合,进行亚结构富集分析并计算预测的基于外部签名集的目标。研究表明,在被埃博拉感染的人细胞中,肯帕洛酮逆转表达的最高预测分子以剂量依赖的方式减轻感染,而不会在两种细胞系中引起细胞毒性。我们还预测了受影响的靶基因和细胞信号通路,这些信号通路和细胞信号通路指向由CDK1-2和GSK3B通路的抑制驱动的免疫应答基因,并可能激活STAT信号传导。但是,在转用埃博拉动物模型之前,应在更多的细胞环境中对肯保隆进行测试。由于显示肯帕龙可诱导协助人类细胞抵抗埃博拉病毒感染的免疫反应,因此这种小分子可能会在阻止其他病毒扩散方面发挥积极作用。 kenpaullone示例只是LINCS L1000数据和基于Web的L1000CDS2工具的冰山一角,因为许多其他应用程序正在等待发现。
五、 材料和方法
5.1 计算LINCS L1000特征方向签名 Computing LINCS L1000 characteristic direction signatures
从l incscloud.org 和GEO(GSE70138)下载了LINCS L1000 3级规范化数据和5级中级Z分数(MODZ)数据。使用定制版的CD方法,使用MATLAB(美国马萨诸塞州纳提克市)对归一化的数据集进行处理。与同一板上的所有对照复制品相比,计算出每个实验复制品的CD单位向量。通过对重复实验中的CD进行平均,可以为每种实验条件计算CD签名。重复之间CD之间的成对余弦距离的平均值用作检验统计量,以评估CD签名的重要性。具体而言,将平均值与由无关CD复制的随机采样构成的零分布进行比较,以计算P值。使用随机乘积算法计算差异表达的基因。CD签名和相关的元数据存储在MongoDB(纽约,NY,美国)数据库中,可从 L1000CDS2 网站下载。
5.2 使用适度的Z分数签名对特征方向签名进行基准测试 Benchmarking characteristic direction signatures with moderated z-score signatures
剂量重要性相关 Dose-significance correlation
CD P值的分布和MODZ强度的分布(distil_ss)绘制为直方图,表示来自LINCS L1000 LJP5和LJP6子集的8,301个独特的L1000实验。 计算了P值与强度之间的相关性,并在散点图中绘制了图形。 在8,301个实验中,确定了1,415个独特条件(如果不考虑剂量)。 对于这些条件中的每一种,都计算出剂量与P值之间以及剂量与MODZ强度之间的相关性。
配体分类
从LINCS L1000 LJP4批次中用配体处理过的癌细胞系中创建的1,374个签名中选择了262个最重要的签名。重要签名由CD签名的重要性确定。计算这些相同有效签名的CD签名之间的成对余弦距离和MODZ签名之间的成对欧几里德距离,并将其组织为两个距离矩阵。多维缩放应用于每个矩阵以可视化散点图中的签名。On the plots ,每个标记都通过其已知的配体类成员,扰动类型,时间点或细胞系进行着色。从两个距离矩阵中提取了两个仅包含使用MCF7细胞系的重要特征的子矩阵。将具有不同距离度量的分层聚类应用于这些子矩阵以生成树状图。树状图的叶子用配体名称标记,并用已知的类成员,扰动类型或时间点着色。
化学-亚结构/表达-特征相关
从LINCS L1000数据中选择了在10μmM下进行6或24 h的小分子处理后,对9种核心细胞系(HA1E,VCAP,HCC515,PC3,A375,HEPG2,HT29,MCF7,A549)进行的实验。这些实验涵盖了20,412个小分子化合物。通过仅保留使用CD签名P值的最重要的唯一签名(细胞系,时间点,小分子和剂量)进一步过滤实验。为了计算这些过滤实验的基因表达签名之间的相似性,余弦距离用于CD和MODZ签名。欧几里德距离也用作MODZ签名之间相似性的另一种度量。检索以简化的分子输入线输入系统(SMILES)格式编码的20,412个小分子化合物的2D化学结构,并将其转换为166位MACCS指纹或ECFP4。谷本相似度用于量化指纹相似度,以谷本系数大于0.9作为截止值。使用Pearson相关分析,将子结构之间的相似性与基因表达签名之间的相似性进行了比较。
药物靶标直接PPI的差异表达
将以SMILES格式编码的LINCS L1000数据中的小分子化合物的化学结构与DrugBank v4.3.31中的化学结构进行了比较。鉴定出297种经FDA批准的药物,其中765种具有至少一个已知靶标。这些靶标的直接已知PPI是从基于文献的哺乳动物PPI网络中收集的,该网络覆盖了9384种蛋白质之间的50478 PPI,如参考文献32中所述。 根据以下标准,从LINCS L1000数据中提取了使用765种具有已知靶标的药物的MODZ和CD签名:(1)MODZ签名牢固,可重现且自连接,如MODZ中的is_gold字段所示元数据; (2)实验的CD签名必须具有显着性,并且如上所示计算P⩽0.05。根据每种药物的差异表达的绝对值对每种CD或MODZ签名中的基因进行排序,然后使用随机游走法将这些等级与该药物靶标的已知直接PPI进行比较。总体平均步行路程绘制在折线图上。
使用来自GEO的独立数据集预测药物目标
为了对使用LINCS L1000基因表达数据的CD或MODZ方法预测药物靶标的能力进行基准测试,我们检查了从GEO挑选的451个单基因摄动基因表达特征,其中摄动基因是至少一种药物的已知靶标, L1000测定。首先通过组合细胞系,剂量和时间点将LINCS L1000药物扰动数据的特征分组。根据重现性(is_gold)和签名强度(distil_ss)选择给定药物最强的签名。为了使用CD或MODZ计算的LINCS L1000的签名对GEO签名进行排序,我们以LINCS L1000签名和GEO签名在所有共享基因空间中的余弦相似性绝对值的递减顺序对GEO基因表达签名进行了排序每个LINCS L1000签名。记录来自Drugbank v4.333的真实药物靶标等级,并将其按比例缩放至来自GEO的单一药物靶标干扰总数。绘制等级等级的累积分布,以评估来自GEO的基因扰动图和LINCS L1000的药物诱导特征的靶标之间的匹配。
。。。。。
参考资料
- L1000CDS2: LINCS L1000 characteristic direction signatures search engine. NPJ Systems Biology and Applications, 2016. https://www.nature.com/articles/npjsba201615
