【4.7.3.6】结构比较--CAD-score(残基-残基接触面积差)
针对天然结构评估蛋白质模型对于蛋白质结构预测方法的开发和基准测试至关重要。尽管迄今为止已经提出了许多评估分数,但是模型评估的许多方面仍然缺乏所需的鲁棒性。在这项研究中,我们提出了CAD评分,这是一种新的评估功能,可以量化模型中的物理接触与参考结构之间的差异。新分数使用了由Abagyan和Totrov提出的残基-残基接触面积差(CAD,contact area difference)的概念(J Mol Biol 1997; 268:678-685)。接触面积是评分的基础,是使用Voronoi细分的蛋白质结构得出的。新引入的CAD评分是一项连续功能,被限制在固定范围内,没有任何任意阈值或参数。用于处理残基的内置逻辑允许对任何完整性程度的模型进行一致的排名。我们在大量不同的模型上测试了CAD得分,并将其与GDT-TS(一种广泛接受的模型准确性度量)进行了比较。与GDT-TS相似,CAD评分在单域蛋白(single-domain protein)上表现出强大的性能,但对物理更真实的模型表现出更大的偏爱。与GDT-TS不同,新分数显示了域重排的平衡评估,消除了对单域,多域和多亚基结构进行不同处理的必要性。而且,CAD评分使直接评估域间或子单元间接口的准确性成为可能。另外,该方法为基于叠加的模型聚类提供了另一种方法。 CAD分数实现既可以作为Web服务器使用,也可以作为独立的软件包在 http://www.ibt.lt/bioinformatics/cad-score/ 上获得。
一、前言
针对实验确定的蛋白质结构(参考)有效评估蛋白质结构模型是蛋白质结构预测方法开发和客观比较的核心。 如果模型中的氨基酸与参考结构一一对应,应该使这项任务变得微不足道。 但是,这种印象是误导的。 这项任务很复杂,尽管多年来已经设计了许多评估评分,但它仍然是研究的活跃领域。
最早,最著名的分数之一是均方根偏差(RMSD,root mean square deviation ):
- RMSD表示两个蛋白质结构中相应的原子在最佳刚体叠加后的平均距离。通常是针对Ca原子计算得出的,但可以应用于残基原子的任何子集。
- 尽管RMSD是一个受欢迎的评分,但只有在差异相当小且分布相当均等的情况下才有意义。
RMSD缺点:
- RMSD的主要缺点是它对较大的局部偏差敏感。甚至很少建模不良的残基,可能对结构和/或生物学的重要性不高(例如,结构不良的蛋白质末端或柔性环),可能会对所得RMSD得分产生重大影响。
- 如果不同的模型包含不同数量的残基,则相应的RMSD值可能会完全误导模型的真实准确性。
尤其是,在早期的CASP实验中,RMSD不足以评估和排名非常不同的蛋白质模型(通常是不完整的蛋白质模型),这是很明显的,该试验是为监测蛋白质结构预测的最新技术而建立的。
因此,CASP实验表明需要在广泛的模型准确性和完整性范围内都具有稳健性的分数。
-
全球距离测试(GDT,Global distance test)是克服RMSD缺点而开发的分数之一。 GDT可以识别模型残差的最大子集(以其Ca原子表示),这些子集可以在特定距离阈值下与参考结构中的相应残基叠加。总体模型准确性由GDT总分(GDT-TS,GDT total score)总结,GDT-TS是通过对在1、2、4和8Å距离阈值下四个独立叠加中获得的残基分数取平均值而得出的单个值。由于严格度不同的多个叠加,GDT-TS能够在广泛的精度范围内对模型进行有效地排名。与RMSD不同,GDT-TS奖励模型的正确的点,而不会为不正确建模的区域增加任何代价。结果,基于GDT的基准测试促进了不仅试图构建最准确的方法,而且试图构建最完整的结构模型的方法。其他分数类似于GDT-TS,包括MaxSub和TM分数。
-
就像GDT-TS一样,MaxSub的目的是识别可以在特定距离阈值下叠加的最大残基子集。但是,与GDT-TS相比,MaxSub仅使用单个3.5Å距离阈值。这使得Max-Sub在排序模型中显得不那么健壮,尤其是那些精度较低的模型。
-
TM得分考虑所有相应的残基对。它使用了与距离有关的加权方案,该方案减少了残差对明显偏离的影响。另外,距离相关的权重随蛋白质大小而变化,与GDT-TS或MaxSub相比,分数的大小相关性更小。但是,类似于MaxSub,TM分数是从单个叠加中得出的。当大小相关性不成问题时(例如,针对同一参考评估模型),GDT-TS中实现的多个叠加具有明显的优势。
毫不奇怪,在CASP实验期间,GDT-TS实际上已成为基于参考的自动模型评估中的核心评分。但是,尽管基于GDT的评分方法很普遍,但它并非没有缺点:
- 由于GDT-TS基于刚体的叠加,因此它在多域蛋白上的表现较差。相互域方向(mutual domain)的轻微变化在生物学上可能无关紧要,但可能会严重影响GDT-TS得分。
- GDT的另一个缺点是它仅使用Ca原子,因此缺少有关残基侧链建模正确性的信息。但是,这是基准测试高精度比较模型或蛋白质结构提纯方法的重要组成部分。
- 另一个也许是最令人不安的问题是GDT-TS得分与蛋白质模型的理化特性之间缺乏直接关系。具有不切实际的特征(例如广泛的原子间碰撞或系统的结构变形)的模型可能仍会获得良好的GDT-TS评分。同样的局限性是类似的得分,MaxSub和TM得分的特征。
为了解决这些问题中的一些,已经提出了对GDT-TS分数的许多修改。 其中一些是针对更高精度模型的更好分辨率。 因此,更严格的GDT-TS版本GDT-HA使用的距离阈值是GDT-TS的一半。 GDC是另一种经过修改的评分,能够包含不同的阈值和残基原子的不同子集。为了使评分牢记空间冲突,建议将排斥项包含在GDT-TS中。 但是,每次修改仅解决了GDT-TS分数的几个限制之一。
因此,显然需要具有GDT-TS的最佳功能(在较宽的模型准确度范围内具有稳健性,并且具有比较不同完整性程度的模型的能力),并具有更有意义的蛋白质结构表示形式。球状蛋白质折叠成特定的3D结构,该结构由残基-残基相互作用定义,并通过物理接触反映出来。因此,似乎接触可能非常适合量化模型相对于参考结构的偏差。此外,接触的比较不需要与所有相关的注意事项进行结构叠加。确实,已经提出了许多使用残基-残基接触概念的分数。但是,通常,这些分数中的“接触”由Ca,Cb或任意指定阈值内所有原子之间的距离表示。显然,在这种分数中接触者的物理意义已经丧失。如果每个残基(例如Ca)仅使用单个原子,那么重要的结构细节也将丢失。
Abagyan和Totrov提出了一个有趣的想法,即使用物理残基-残基接触的显式描述进行模型评估。 他们建议使用残基-残基接触面积作为比较模型和参考结构的基础。 此外,他们引入了单一数字评分,即接触面积差(CAD,contact area difference),以衡量整体模型的准确性。 Abagyan和Totrov定义的CAD具有许多吸引人的功能。 它是连续的且无阈值,可在各种模型精度下工作,可以充分惩罚域,片段和侧链重排,并捕获蛋白质结构的基本几何特征。
但是,原始CAD具有一些特性,使其无法用于大规模评估方法(例如CASP实验)。
- 首先,CAD仅考虑模型和参考结构共有的残基。 这意味着将针对完整的参考结构评估完整的模型,而针对相应的参考片段评估建模的短片段。 换句话说,参考的确切选择取决于模型的完整性。 几乎不能认为这是对不同方法进行基准测试的客观模式。
- 其次,归一化CAD术语不仅包括参考结构而且还包括模型的残基间接触区域。 尽管预计这不会对总分产生很大影响,但仍然使CAD归一化模型特定。
在这里,我们引入了一个新的基于接触面积差异的评分(CAD-score),用于针对参考结构进行模型评估。 它结合了原始的CAD概念和GDT-TS评分设计的一些潜在思想。 新的CAD分数使用更精确的算法进行接触面积计算。 此外,新算法可在原子水平上解析残基与残基的接触,从而有可能单独考虑残基原子的子集(主链,侧链)。 CAD评分具有一种处理模型中残基的方法,因此,与GDT类似,它可以有效地对完整和不完整的模型进行排名。 新的CAD分数针对参考结构中的触点进行了归一化,并且始终在0到1之间。CAD分数的计算和可视表示软件,接触图和局部偏差都可以通过Web获得。 服务器并作为独立程序包。
二、材料和方法
2.1 残基-残基接触面积的计算
残基接触面积是使用蛋白质结构细分(tessellation)方法得出的。 我们使用称为3D球体的Voronoi图(也称为加法加权Voronoi图或Apollonius图)的细分。 这种细分中的球体对应于范德华半径的重原子。 在这里,我们将范德华半径用于Li和Nussinov衍生的蛋白质重原子。 对于每个原子,我们可以定义Voronoi cell,这是一组比所有其他原子更接近此特定原子的点。 蛋白质结构的Voronoi图是蛋白质所有重原子的Voronoi细胞集合。 如果两个原子的Voronoi cell共享一个共同的点子集,则称两个原子为Voronoi邻居。
原子间接触是基于McConkey等人提出的想法从原子的Voronoi图得出的。 如果水分子不能容纳在它们之间,则相邻的蛋白质原子被定义为相互接触。 因此,原子的完整接触表面由半径等于球的范德华半径与水分子的标准半径(1.4Å)之和的球体表示。 我们称其为接触球。 如果满足以下两个条件,则原子i的接触球面上的点p属于与原子j的接触面:(1)i和j是Voronoi邻居; (2)p比i或i的任何其他邻域更靠近j。如果邻位环邻氨基甲酸酯,则该分子属于可接触溶剂的表面。
对于给定的原子,我们通过与对应于相邻Voronoi细胞交界处的双曲面相交的原子接触球的三角表示来构造其接触表面。 结果,原子的整个接触球明确地划分为接触区域和溶剂可及区域。 残基-残基接触是通过简单地将相应残基原子之间的接触分组来构造的。 不考虑形成顺序相邻的残基的肽键的C和N原子之间的接触。 由于接触是在原子水平上解析的,因此我们不仅可以为整个残基定义接触,还可以为其原子的各个子集(例如主链和侧链)定义接触。 图1说明了Voronoi细胞和接触球的组合如何用于构造原子[图1(A)]和残基[图1(B)]的接触表面。
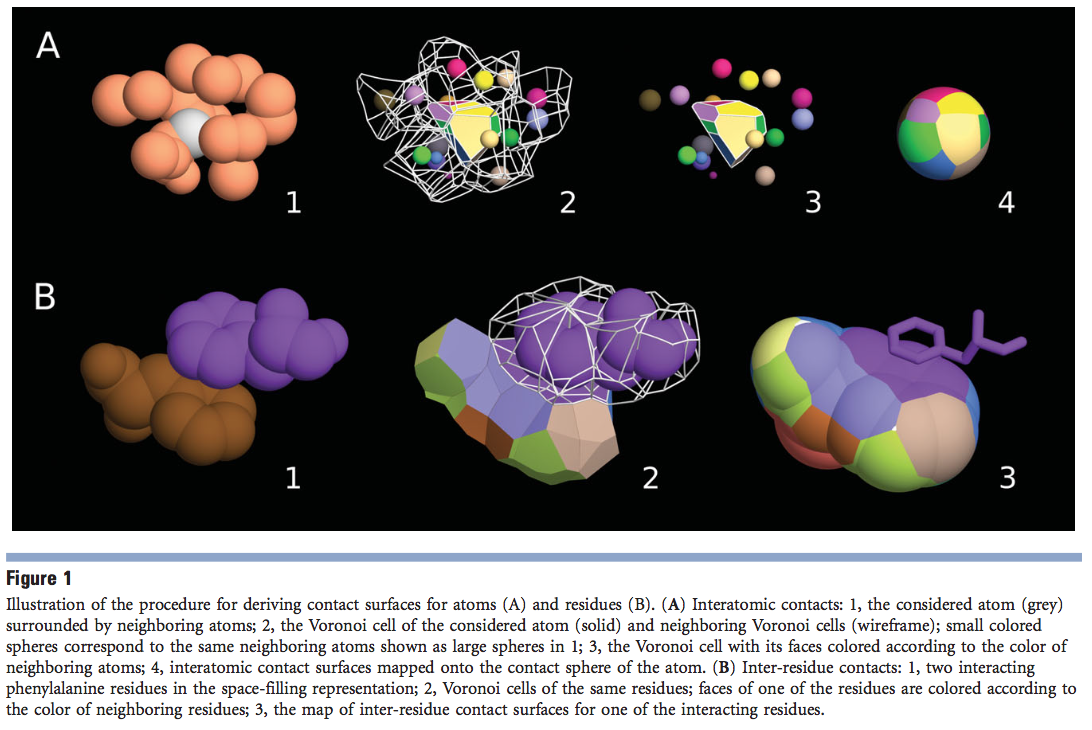
使用为Voroprot开发的算法的修改,可以计算出蛋白质结构和原子间接触区域的细分。 改进的算法(在其他地方有详细说明)的特征是速度的显着提高,这是通过优化空间搜索操作来实现的。 另外,对各种输入异常的改进处理使修改后的算法可以处理甚至具有物理上不切实际的特征的蛋白质结构。
2.2 CAD分数定义
我们基于以下三个主要考虑因素定义了CAD评分:
- 应根据参考结构(目标)中的接触面评估模型中的接触面; 2。 对模型中任何遗漏的残基的处理方式应与正确预测其所有接触方式相同;
- 特定接触面的强烈过度预测(非物理重叠)应等同于完全失去该接触。 CAD分数的数学定义如下所示。
令G表示目标结构中具有非零接触面积T(i,j)的所有残基对(i,j)的集合。 然后,对于每个残基对(i,j)∈ G,我们计算模型中的接触面积M(i,j)。 如果模型中目标中不存在其他残基,则这些残基将从接触面积的计算中排除。 如果目标中存在一些残基,但模型中缺少该残基,则将模型中该残基的所有接触面积分配为零。
然后,对于每个残基对(i,j)∈ G,我们可以将接触面积差定义为目标T和模型M中残基i和j之间的接触面积的绝对差:
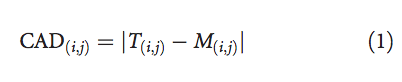
为了对接触区域的过高预测和过低预测进行对称处理,我们使用定义为以下条件的 bounded CAD(i,j)代替原始CAD(i,j)值:
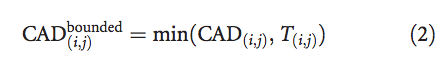
整个模型的CAD分数定义为:
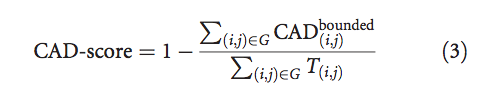
等式(3)的分子之和永远不会超过目标结构中所有接触面积T(i,j)的总和。 换句话说,由等式(3)定义的CAD分数始终在[0,1]范围内。 如果模型和目标结构相同,则CAD分数=1。在另一个极端,如果没有以足够的精度复制单个接触(没有满足以下条件的情况:CAD(i,j)<T(i,j )),CAD-score = 0。
2.3 CAD分数定义
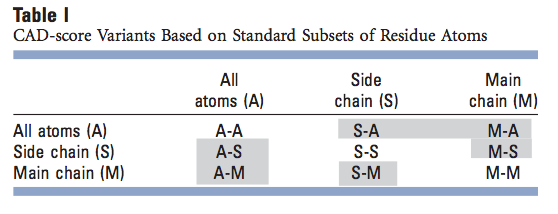
我们的算法以单个原子的分辨率计算残基间接触面积。因此,我们不仅可以定义整个残基的接触面积,还可以定义其原子的任何子集的接触面积差。在所有情况下,均以存在所有原子的方式计算接触面积,但如果考虑残基原子的子集,则仅保留与该子集对应的接触面积。在这里,我们使用所有残基原子(A)和原子的两个标准子集,即主链(M)和侧链(S),得出9个CAD分数变体(表I)。三对CAD分数变体(A-S和S-A,A-M和M-A,以及S-M和M-S;表I中的灰色背景)不完全对称。例如,甘氨酸没有侧链,因此不能形成任何S-A接触,但可以形成A-S接触。但是,出于实际目的,这三对CAD分数变量可能被认为是多余的。结果,对于残基原子的标准子集,有六个非冗余的CAD分数变体,可用于解决针对参考结构评估模型的不同问题。
三、结果
为了测试CAD评分的特性及其在评估和排名模型中的有效性,我们将其应用于在CASP9(第九个社区范围内的蛋白质结构预测方法评估)期间获得的模型。 CASP模型是通过大量不同方法生成的,因此代表了广泛的精度。另外,该集合包含不同完整性程度的模型,包括完整模型,缺少一些残基和仅短结构片段的模型。此外,模型的物理合理性差异很大。其中一些具有结构特征,使人联想到高分辨率的实验结构,而另一些则具有许多不切实际的特征,例如空间碰撞和强烈偏离共价键的几何形状。 CASP模型的所有这些方面使它们成为基于基于参考的自动模型评估得分的出色测试集,因为该集对客观和公正的模型排名提出了严峻挑战。为了具有代表性且冗余最少的集合,我们仅考虑了由自动方法(服务器)生成的CASP9模型,对于给定的预测目标,每种方法采用一种最自信(第一)模型。由于CAD评分是一种全原子量度,因此我们从以简化或不完整形式表示氨基酸残基的方法所产生的分析模型中排除了这一点。还排除了其中一个靶标(T0629; T4噬菌体的长尾纤维蛋白gp37)的模型。 T0629形成针状平行均聚物,考虑到分离出的单链在结构和生物学上均无意义。
3.1 CAD分数是用于评估和排名单域模型( single-domain models)的可靠方法
第一步,我们决定将CAD评分与GDT-TS(一种经受时间测试的标准CASP评分)进行比较,通常被认为是最有效的基于参考的评分。 为了对CAD评分和GDT-TS进行总体比较,我们针对评估者定义的预测目标的各个域(更准确地说是“评估单位”)选择了CASP9模型。 对于由此产生的8429个多样化模型集,我们汇总了GDT-TS和CAD得分。 GDT-TS值来自Prediction Center的数据档案库( http://www.predictioncenter.org/ ),而CAD分数的不同变体则如“材料和方法”中所述进行了计算。 图2显示了GDT-TS与六个非冗余CAD分数变体之间的关系船。
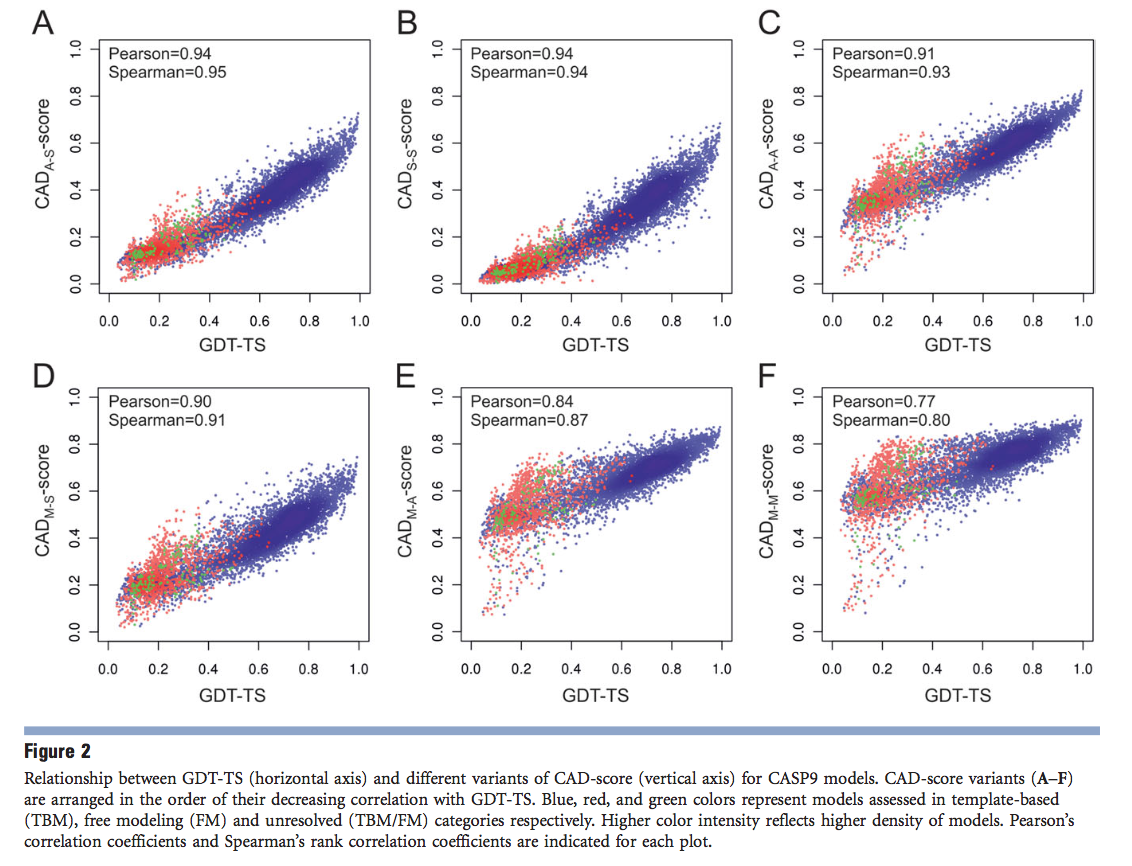
显然,GDT-TS和CAD得分值之间存在很强的相关性,考虑到分数的不同性质,这令人惊讶。值得注意的是,不仅对Pearson的相关系数如此,后者取决于两个分数之间的线性关系。在所有情况下,Spearman的排名相关性都可以获得更好的值,这表明GDT-TS的排名与CAD评分的排名在多大程度上相符,而无需假设两个评分之间存在线性关系。特别是,三种类型的CAD分数(“所有原子-侧链”(AS),“侧链-侧链”(SS)和“所有原子-所有原子”(AA))显示出最强的相关性[图2(A–C)]。对于这三个CAD得分变体,Pearson的相关系数在(0.91-0.94)范围内,而Spearman的等级相关值在(0.93-0.95)范围内。其他三种CAD评分,特别是基于“主链-主链”(M-M)接触的变量,其相关性较弱。我们认为,很大程度上,较低的相关性可能由未与结构的全局拓扑链接的局部M-M联系人确定。如果是这样,那么二级结构的类型应该是主要因素。确实,当单独分析时,富含b链的蛋白质(许多非局部M-M接触)的相关性得到改善,而对于a螺旋蛋白(主要是局部M-M接触),其相关性进一步降低(支持信息图S1)。
我们还研究了CAD评分与GDT-HA之间的相关性,GDT-HA是更为严格的GDT-TS变体。 GDT-HA类似地从四个独立的叠加中得出,但是它们的阈值距离(0.5、1、2和4Å)仅为标准GDT-TS的一半。 因此,GDT-HA可以为精度更高的模型提供更好的分辨率。 关联性最好的CAD分数变体相同(A-S,S-S和A-A),并且它们的相关值仍然非常相似。 即,皮尔逊相关系数和斯皮尔曼相关系数的范围分别为(0.91-0.95)和(0.92-0.95)(支持信息图S2)。
CAD评分中使用的唯一可调参数是蛋白质原子的范德华半径(VDW)半径的值。 由于文献中已经报道了不同的VDW半径集,因此我们询问结果是否对特定集合的选择敏感。 为此,除了使用Li和Nussinov报告的使用标准VDW半径评估CASP9模型之外,我们还使用同一作者得出的最小VDW半径集重复了分析。 尽管两个VDW集之间的差异是可变的,并且有些差异相当显着(最高0.45Å),但我们仅观察到CAD得分值及其与GDT-TS或GDT-HA的相关性可以忽略不计(支持信息表S1) 。 毕竟,这个发现应该不会太令人惊讶,因为CAD评分:一种新的模型精度度量CAD评分是基于接触面积差异而不是绝对接触面积大小。
综上所述,这些分析揭示了CAD评分对单域蛋白的强大性能。 特别是,三种CAD评分类型(A-S,S-S和A-A)脱颖而出。 它们提供了一些最高分辨率和与GDT-TS / GDT-HA的最佳关联。 因此,我们将主要集中在这三个CAD分数变体的属性上。
3.2 CAD分数可促进结构模型的物理逼真度
通常假定较好的模型得分表示参考结构的更准确表示。 但是,已经注意到,包括GDT-TS在内的一些模型评估分数对不切实际的结构特征(如空间碰撞或残渣几何形状的偏差)相当不敏感。 因此,根据特定分数进行的改进可能会以结构模型的物理真实性为代价。 换句话说,某些蛋白质结构预测方法,尤其是针对特定分数进行了优化的方法,可能会根据该分数“改善”其性能,而实际上并未提高模型的准确性。
CAD分数呢?根据CAD评分对模型的改进如何与它们的物理真实感相关联?由于CAD评分与GDT-TS高度相关(图2),在这方面与GDT-TS相比,它的表现如何?为了回答这些问题,我们分析了CAD评分和GDT-TS排名冲突的模型对,即CAD评分和GDT-TS为所考虑的模型中的不同模型分配了更好的值。我们询问在这些情况下哪个得分与模型的物理真实性更一致。我们选择了MolProbity分数来衡量物理真实感。 MolProbity是广泛使用的结构质量评估套件之一。 MolProbity得分是一个单一数字,代表从大量高质量蛋白质晶体结构中收集的中心MolProbity蛋白质统计信息。得分考虑了非键合原子之间的冲突,受偏爱区域之外的骨架Ramachandran构象以及侧链旋转异构体离群值。与GDT-TS和CAD-分数不同,MolProbity分数不是以参考为中心的度量。它并不能说明模型与本地结构的接近程度。相反,它报告模型的“蛋白质样”(protein-like)状态。因此,MolProbity可被视为解决两个基于参考的得分之间排名冲突的独立“判断”。
我们将分析限于单个域(评估单位)的合理准确的模型,因为考虑完全不正确的模型的物理现实性将毫无意义。因此,我们选择了GDT-TS阈值超过0.6(60%)的模型,并编译了GDT-TS与这三个CAD评分变体之间的排名冲突的模型对。然后,我们研究了MolProbity得分如何对同一对中的模型进行排名。分析结果表明,在排名冲突的情况下,MolProbity评分支持的CAD评分要比GDT-TS强得多[图3(A)]。在这三个CAD评分变体中,CADA-A得到了最大的MolProbity支持,其次是CADA-S,然后是最严格的CADS-S。但是,可能期望对MolProbity,GDT-TS或CAD得分值有微小差异的模型对会在结果中产生一定程度的噪声。因此,我们进行了两个附加测试,旨在逐步消除噪声的影响。首先,我们只看了那些有冲突的排名,在所有考虑的模型上,它们的绝对MolProbity得分差异大于MolProbity得分分布的标准偏差[支持信息图S3(A)]。结果,与Mol-Probity的CAD评分协议显着增加[图3(B)]。对于第二项测试,除了限制MolProbity得分差异外,我们要求GDT-TS或CAD-得分值的差异也应大于相应的标准偏差[支持信息图S3(B–E) ]。第二项测试进一步强调了对CAD分数的压倒性的MolProbity支持[图3(C)]。例如,CADA-A的排名与25个案例中的24个案例的MolProbity分数相符,而只有一种情况对GDT-TS成立。总体而言,这些分析表明,如果对模型的相对排名存在分歧,则CAD评分会比GDT-TS赋予物理上更真实的模型更好的评分。此CAD评分属性可能与诸如对模型进行较高准确度排名和评估模型改进之类的任务特别相关,因为根据CAD评分获得更好的性能也将强烈暗示物理现实性的改善。

3.3 CAD评分消除了将多域蛋白拆分为域以进行模型评估的必要性
许多蛋白质由多个结构域组成。但是,GDT-TS和其他基于刚体叠加的分数(例如TM分数,RMSD)对即使很小的域方向差异也很敏感。结果,整个结构的模型分数常常与单个领域的模型分数脱节。通过将目标结构划分为多个域并执行基于域的评估,可以解决该问题。但是,由于没有通用的域定义标准,因此通常无法明确定义域的数量及其确切边界。此外,并非总是很清楚是否有必要将多主体目标结构划分为用于评估目的的域。 Grishin及其同事最近引入了一种简单的方法来帮助确定是否需要划分域。他们的方法(在“官方” CASP9评估中使用)基于对整个链模型的GDT-TS得分与各个域的GDT-TS加权总和之间的相关性分析。加权总和的定义如下:将每个域的GDT-TS得分(与域的长度相乘)相加,然后除以域长度的总和。主要思想是,如果整个域的得分链模型在系统上低于(或高于)领域得分的加权和,因此应考虑划分为领域的情况。由于这个想法很笼统,因此我们决定基于CAD评分进行类似的分析,并将结果与GDT-TS获得的结果进行比较。但是,与单个结构域的总和相比,某些CASP9全链靶结构具有额外的残基。为了使分析完全客观,我们从多结构域全链目标结构中删除了这些额外的残基,以使全链结构和结构域总和具有完全相同的残基。然后,我们通过CAD评分和GDT-TS针对这些全链目标评估了模型。使用LGA重新计算了后者的数据。图4显示了针对24个多域目标的1287个模型的结果分析。GDT-TS图之间存在明显的差异。和基于CAD分数的图[图4(B–D)]。对于GDT-TS,本质上对于所有模型,域得分的加权总和高于整个结构的得分。这重申了评估者做出的选择,以将这些CASP9目标解析为域(评估单位),以便使用GDT-TS进行健壮的模型评估。与GDT-TS相比,所有三个CAD分数变体[图4(B–D)]最多只显示出全链结构得分与域组合得分之间的微小差异。换句话说,基于CAD评分的评估甚至可以在不将结构解析为域的情况下,对多域蛋白的模型进行客观比较。

3.4 CAD评分可在多域蛋白质模型中平衡评估域间排列的准确性
尽管CAD评分显示基于域的评估与全链评估之间几乎没有差异(图4),但重要的问题是这是否反映了域重排的适当评分。我们认为,预测相互域排列的准确性不应通过域之间方向的简单错误来判断。如果仅通过连接的linker将域保持在一起,则任何固定的相互方向可能在结构和/或生物学上无关紧要(尤其是在linker灵活的情况下)。在这种情况下,不预测晶体结构中观察到的特定取向的代价将是不公平的。相反,如果域共享广泛的interface,则它们的具体排列方式表明结构和/或生物学重要性,因此应对评估得分有更大贡献。换句话说,埋藏在结构域界面上的蛋白质表面积所占的比例越大,它应该能够对模型的总评分施加的潜在影响(正或负)就越大。按照这种逻辑,我们分析了域排列对模型总得分的预期和观察贡献。我们将预期贡献定义为埋在目标的域-域界面中的溶剂可及表面(SAS)的比例,该比例针对给定的全链模型的准确性进行了校正。通过简单地将interface处的SAS分数乘以整个链分数即可进行校正。通过从各个域的SAS的总和中减去整个链结构的SAS,再除以2,可以确定埋藏的SAS。当然,定义域排列的预期贡献是简单的,因为我们认为接口预测的准确性与所有域的平均准确性相同。但是,该概念对于探索预期贡献和观察贡献之间的关系很有用。观察到的贡献定义为全链得分与域得分的加权总和之差(如图4所示)。
我们分析了CAD得分和GDT-TS的域间interface预测组件对模型总得分的预期贡献和观察贡献之间的关系。结果显示在图5中。我们仅包括那些多域蛋白质模型的数据,该模型的所有单个域的GDT-TS值均超过0.4(40%),因此有望代表至少正确的结构折叠。尽管有一些数据噪音,但该图显示出GDT-TS和CAD评分的行为截然不同。
根据GDT-TS的数据[图5(A)],可以得出两个重要的观察结果。首先,对总分的最大观察贡献是最大预期贡献的几倍。这是GDT-TS属性的结果,该属性极大地夸大了域重排,从而使基于域的评估成为必要。其次,对于某些期望值最小的模型,这种夸张最为明显。换句话说,给定单个域的平均质量相似,具有最小域间interface的目标模型更可能对整个链结构产生较差的分数。
相反,对于CAD分数[图5(B–D)],随着预期贡献的增加,观察到的域排列分数对总分数的贡献趋于增加。 最好的一致性由CADA-A得分,随后的CADA-S和CADS-S得分显示。 尽管这种关系有些嘈杂,但是观察到的贡献几乎从未超过预期的贡献,这表明域排列错误对总分的平衡影响。
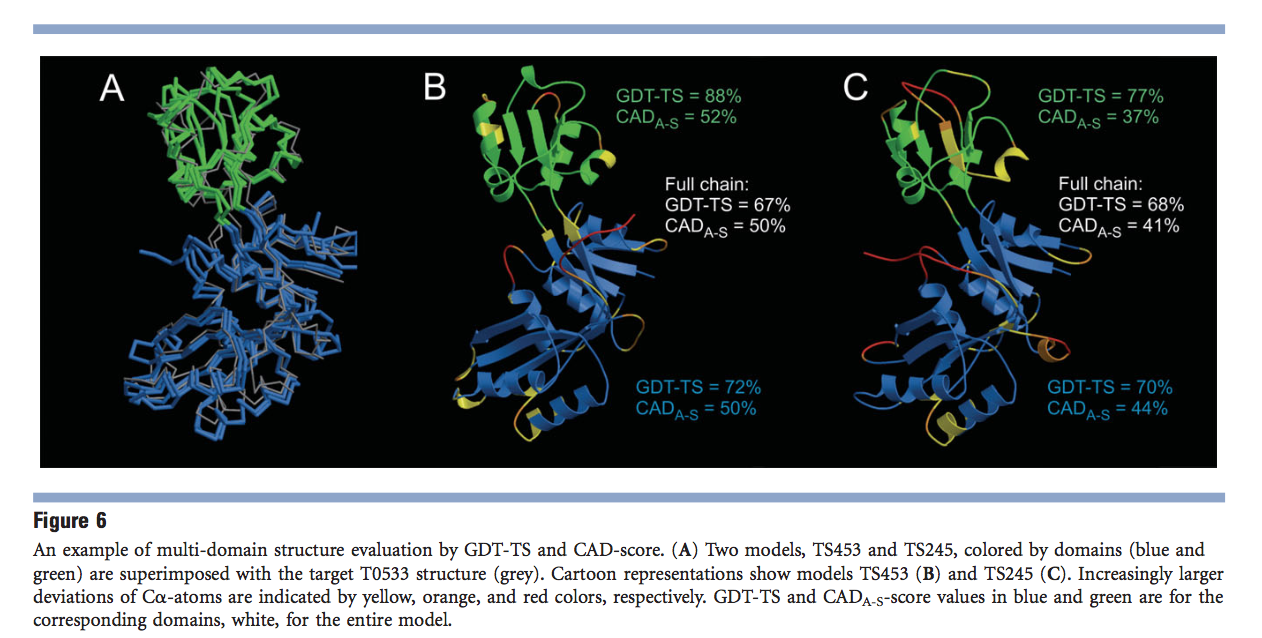
在评估多域目标的模型后,随着CAD评分的消失,GDT-TS问题的说明性示例如图6所示。CASP9模型TS453的两个域的GDT-TS得分[图6(B) )]优于TS245的[图6(C)]。然而,尽管在两个模型中在视觉上非常相似的相互域布置[图6(A)],TS453被分配了更差的全链GDT-TS值。显然,这不能被认为是公正的评估。相比之下,CAD评分不仅为TS453的各个域分配了更好的分数,而且,正如可以预期的那样,还为全链模型分配了更好的分数。即使在同一模型中,GDT-TS倾向于高估相互域排列中的微小差异的趋势也很明显。可以预期,全链模型的准确性介于得分最差的域和得分最高的域之间。但是,根据GDT-TS,图6中的两个全链模型都比其最不精确的域差。同样,CAD分数不存在此问题。
由于域的相互排列在概念上与蛋白质链的排列没有区别,因此CAD评分也可用于评估蛋白质复合物模型的准确性。 亚单位间的界面越大,其预测精度对蛋白质复合物总CAD分数的影响就越大。
3.5 CAD评分可以直接评估域间或子单元间interfaces的准确性
除了为整个多域或多亚基结构评分模型外,CAD评分还提供了一种评估界面预测准确性的直接方法。唯一的区别是评估模型所依据的参考。在这种情况下,参考将被定义为源自不同蛋白质结构域(域间界面)或不同蛋白质亚基(亚基间界面)的残基之间的接触区域。图7提供了不同精度的域间和子单元间接口的特定示例。第一个示例[图7(A)]说明了目标T0533的两个模型的域间接口的准确性,上面已对此进行了详细分析。再次证明,TS453模型比TS245具有更准确的域间接口。另一个示例[图7(B)]的特征是目标T0576的两个低聚预测中具有不同准确性的亚基间界面。 CAD评分将TS458识别为两个模型之一,它具有针对该目标的最准确的界面。该CAD分数分配完全符合CASP9对低聚预测的评估。

3.6 CAD评分Web服务器和独立软件
为了使计算CAD分数的方法易于使用,我们将其实现为Web服务器,网址为 http://www.ibt.lt/bioinformatics/cad-score/ 。 该服务器具有简单直观的界面。 有几个主要功能。 CAD评分服务器可以针对参考结构评估单链蛋白质模型以及蛋白质复合物(多链结构)模型的准确性。 另外,服务器可用于专门评估接口预测的准确性。 该接口可以以非常灵活的方式定义。 它可以在不同的蛋白质链之间定义,也可以在任何用户定义的残基范围之间定义(如在蛋白质结构域之间的界面中)。
服务器的输入是单个模型或多个模型,以及要对其进行评估的参考结构。 CAD评分服务器会计算所有CAD评分变体,但默认情况下仅报告与GDT-TS最相关的三个变量,即CADA-A评分,CADA-S评分和CADS-S评分。作为模型评估结果的摘要,服务器不仅提供CAD分数变体,还提供TM分数软件生成的TM分数,GDT-TS和GDT-HA值。可以通过这些分数中的任何一个对模型进行排序。除摘要外,服务器还会为蛋白质模型生成位置相关的颜色编码的CAD错误配置文件。这些配置文件对于视觉识别一组模型中蛋白质链特定区域中的独特或常见模式特别有用。用户还可以详细浏览各个模型。详细的分析包括模型和参考的叠加接触图,具有不同平滑窗口的误差轮廓以及根据局部CAD误差用颜色编码的模型和参考结构的Jmol可视化。
连同CAD分数Web服务器一起,可以在本地使用相应的独立软件包。 独立的CAD评分软件可计算与服务器相同的数据,但可提供更大的灵活性,尤其是在需要大规模评估或模块集群的情况下。 可以从CAD评分Web服务器地址下载该软件。
四、讨论
蛋白质结构预测方法的开发和用于基准测试的评分是相互依赖的。 稳健而有效的评分可促进蛋白质结构预测方法的改进。 另一方面,模型准确性的整体提高需要更敏感和更全面的评估。 目前,由于结构预测方法的改进和基于模板的模型的优势,焦点已转向骨干以外的结构特征的准确性。 更多的重点放在计算模型的物理合理性上。 在多域蛋白质模型中评估相互域排列的准确性以及蛋白质复合物中亚基排列的能力也变得越来越重要。
在这项研究中,我们提出了CAD评分,这是一种用于综合评估结构模型的新模型评分功能。 CAD分数基于Abagyan和Totrov最初提出的接触面积差(CAD)概念。 但是,新分数在设计和算法实现上有很大差异。
关键差异之一是模型中缺失残基的处理。原始CAD仅考虑目标和模型共同的残基子集。在这方面,它让人联想到RMSD,后者只能根据一组常见的残基进行计算。在新定义的CAD评分中,未能将残渣包含到模型中以及无法预测其所有接触均得到了相同的对待。换句话说,CAD评分鼓励构建完整的模型。建模不正确的区域最多只能对分数造成微不足道的改善;但是,与根本不建模的情况相比,即使模型的严重错误区域也不会使得分更糟。在这方面,CAD评分的设计与GDT-TS相似,虽然没有奖励,但同时也不会惩罚严重不准确的区域。我们认为,这是基于参考的模型评估得分的非常积极的特征,因为它可以测试蛋白质结构预测中的新大胆想法,而不会因局部误差大而受到惩罚。
第二个区别是规范化(normalization)过程。对于相同参考结构(目标)的不同模型,由Abagyan和Totrov提出的CAD编号中的归一化因子是不同的。这使得给定目标的模型排名存在问题。在我们的案例中,归一化术语对于给定目标是恒定的,无论异常模型或评估模型有多不同。
另一个区别是值的范围。最初建议的CAD编号不能总是保证落在0到1(0-100%)的范围内。相反,新定义的CAD分数永远不能超出[0,1]范围。对于给定的残基对,最大接触面积差的“对称”边界可以确保这一点。我们以无法预测现有contact的“强烈”过度预测为准。 “绝对”过高预测是指绝对接触面积差大于参考接触面积本身的情况。在两种极端情况下,我们都认为预测同样是错误的,因此接触面积差受参考接触面积的限制。结果,模型的有界接触面积差之和永远不会超过目标的接触面积之和。
在我们的案例中,得出接触面积的算法与原始CAD研究也存在很大差异。 我们使用蛋白质结构细分方法得出接触面积。 它使我们能够考虑周围残基对中其他残基的影响。 在最初的CAD研究中,一对残基的接触面积是独立计算的,因此往往会高估接触面积的大小。 另外,在两种方法中接触面积的分辨率是不同的。 与Abagyan和Totrov相比,我们在重原子水平上计算接触面积,这使我们不仅可以得出整个残基的接触面积,还可以得出残基原子子集(例如主链和侧链)的接触面积。 反过来,这使我们可以定义许多CAD分数变体,以解决模型准确性的不同方面并提供不同程度的敏感性。
在这项研究中,我们探索了新引入的CAD评分的属性,并将其与GDT-TS(主要是基于参考的模型评估广泛接受的评分)进行了比较。我们发现,对于单个结构域,CAD得分与GDT-TS(图2)和GDT-HA(支持信息图S2)显示出很强的相关性。在这两种情况下,对于那些包含所有残基原子或侧链的任何组合的CAD分数变体,都获得了最强的相关性。似乎所有原子和侧链之间的接触(A-S)甚至侧链之间的接触(S-S)与GDT-TS的关联都比所有原子与所有原子之间的接触(A-A)更好。然而,侧链约占蛋白质结构的三分之二,而且显然它们的堆积是产生特定折叠模式的原因。仅在接触的至少一侧包含主链原子的CAD分数变体显示出较弱的相关性,主链与主链(M-M)变体占据了较低端。主链与主链接触的特性根据二级结构类型而显着不同。虽然在β-sheets中,这些接触是由全局拓扑定义的,但对于a螺旋,它们是局部的,并且主要由二级结构分配的精度定义。显然,α-螺旋结构内缺乏非局部接触是使M-M变体与GDT-TS的相关性最小的一个主要因素(支持信息图S1)。
与GDT-TS和其他基于结构叠加的方法相比,CAD分数的重要优势之一是对多域蛋白质和蛋白质复合物模型的鲁棒性评估。 我们的分析表明,与GDT-TS相比,单个域的CAD得分与全链结构紧密相连(图4)。 此外,域间或子单元间interface的准确性是总分的重要组成部分。 界面越广泛,对其总评分的潜在改善或降低的可能性就越大(图5)。 尽管完全有可能进行基于域的模型评估,但CAD评分消除了将结构切分成域以获得有意义的结果的必要性。 此外,即使将结构拆分为多个域,不精确甚至完全错误的域边界定义也不会严重影响CAD评分的性能,这将对基于GDT-TS的评估产生很大影响。
根据CAD评分,模型的准确性仅取决于残基(或残基原子的子集)之间的接触面积与参考结构中的接触面积对应的紧密程度。 但是,看似对模型精度的简单定义实际上包含了许多结构特征,例如原子间距离,二面角,氢键和键长。 使用特定的模型评估得分训练的蛋白质结构预测方法,在某些情况下可能会通过优化某些模型结构参数而以“其他”为代价来“改善”其性能。 在这里,我们表明CAD评分与模型的物理真实性相关联,其比GDT-TS强得多(图3)。 特别是,CAD评分的这一属性可能与评估模型的改进有关,而事实证明这是一个令人惊讶的难题。
尽管我们在开发新的CAD评分时考虑了基于参考的模型评估,但是该方法对于其他任务(例如结构模型的聚类)可能是有价值的工具。模型聚类是许多当前蛋白质结构预测方法所采用的步骤之一,尤其是在没有合适的结构模板的情况下。聚类步骤用于从大量的候选结构(诱饵)中识别出近自然结构。由于残基之间的接触面积直接反映了物理相互作用的强度,与基于RMSD或GDT的基于笛卡尔距离的方法相比,CAD得分值可能更适合于对具有相似能量的模型进行分组。由于聚类通常涉及大量模型,因此聚类方法需要快速。在基于CAD的聚类中,最慢的步骤是计算各个模型中残基之间的接触面积。但是,一旦完成,成对CAD分数的后续计算将非常快。支持信息图S4中提供了使用CAD分数进行模型聚类结果的示例。
CAD分数基于原子间的接触,因此它不仅限于蛋白质结构。 类似的方法可用于评估形成复杂3D结构(例如RNA)的其他生物分子的模型。 同样,对蛋白质-蛋白质界面(域间或亚基间)准确性的评估可以很容易地扩展到蛋白质-配体界面的更一般情况。 显然,基于CAD评分的评估最适合大型界面,例如蛋白质-核酸复合物中的界面,但即使对于蛋白质与小分子之间的界面,也可能具有足够的信息价值
五、总结
新推出的CAD分数具有许多吸引人的特性。 它基于残基之间的物理接触,从而直接反映蛋白质结构内的相互作用。 它是一个连续的无脱粒(threshold-free)函数,可在严格定义的范围内返回定量准确度得分。 CAD分数的定义不包含任何任意参数。 CAD评分提供了一个统一的框架,用于评估准确性和完整性不同的单域,多域甚至多亚基蛋白质结构模型。 虽然CAD得分与GDT-TS在单域结构上高度相关,但CAD得分却更加强调模型的物理真实性。 我们相信,所有这些吸引人的特性使CAD评分成为开发和评估蛋白质结构预测和改进方法以及基于它们彼此相似性的聚类模型的有价值的工具。
参考资料
- CAD-score: A new contact area difference-based function for evaluation of protein structural models
