【2.3】promotif
PROMOTIF–a Program to Identify and Analyze Structural Motifs in Proteins
PDB文件中的HELIX和SHEET的注释来自Promotif,
Helix and sheet records are automatically generated by Promotif software.
我们描述了一套程序PROMOTIF,它可以分析蛋白质坐标文件并提供有关蛋白质结构基序的详细信息。 该程序目前分析以下结构特征:二级结构; β和γ转; 螺旋几何形状和相互作用; β-链和β-折叠拓扑; 贝塔凸起 β-发夹 beta-alpha-beta单位和psi环; 二硫键; 和主链氢键模式(secondary structure; beta-and gamma-turns; helical geometry and interactions; beta-strands and beta-sheet topology; beta-bulges; beta-hairpins; beta-alpha-beta units and psi-loops; disulphide bridges; and main-chain hydrogen bonding patterns)。 PROMOTIF创建后记文件,显示蛋白质中每种基序类型的示例,以及摘要页面。 该程序还可以用于比较一组相关结构中的基序,例如NMR结构集合。
一、前言
蛋白质结构以分层的方式排列,从形成螺旋和链的基本氨基酸构件(一级结构)开始,这些二级结构单元以各种方式组装以形成蛋白质的最终三级结构。尽管现在已知大量蛋白质的结构,但是它们在二级,超二级和三级水平上显示的模式要小得多。在第三级上,已经发现Brookhaven蛋白质数据库(Bernstein等,1977)的相对域折叠相对较少(例如Sander&Schneider,1991; Chothia,1992; Orengo等1993)。在结构层次中较低的水平上,即使在无关的蛋白质中,也经常观察到相对少量的基序。 这些包括二级结构的基本单元 α-和310螺旋,β链,以及β和γ-turns,以及超二级结构,例如β-发夹,β-α-β units,β-sheet拓扑。
由于多种原因,对蛋白质中这些常见基序的位置和结构的更详细的了解很重要。 它可以深入了解蛋白质与其可能的进化起源之间的关系。 这也加深了我们对氨基酸序列与三级结构之间关系的理解,进而可用于通过同源性进行建模,从序列中从头开始预测结构的结构以及新型蛋白质的设计。 超二级结构也是蛋白质折叠中成核位点的良好候选者。 最后,许多基序(例如 β-turns和 β-bulges)在功能上很重要,因为已发现它们涉及活性位点和配体结合表面
由于这些原因,已经对许多这些motifs进行了详细分析(例如,β-发夹[Sibanda&Thornton 1985,1989,Efimov,19871; ] β-转[Wilmot&Thornton 1988,1990; Hutchinson&Thornton,19941;] β-凸起[Richardson,1981; Chan等,1993],)并且已经设计出分类方案来描述它们出现的构象。 显然,现在可用的大量蛋白质结构意味着,对于方法而言,识别和分析此类基序以实现自动化非常重要。 在本文中,我们描述了一套称为PROMOTIF的新程序,该程序可以分析蛋白质坐标文件并提供该蛋白质中结构基序的详细信息。
二、结果与讨论
PROMOTIF的第一个版本提供有关蛋白质以下结构特征的信息:二级结构; β和γ-turns; 螺旋几何形状和相互作用; β链和β-sheet拓扑; β-bulges; β发夹; β-α-β个单位; ψ循环; 二硫键; 和主链氢键模式。 为每种类型的基序创建了后记文件,该程序还生成了一个摘要页面,该页面对蛋白质中发现的每个基序进行了更简短的描述。 大多数主题都是根据已发表论文中定义的规则进行分析和分类的。 下面以蛋白质牛DNASE I(Brookhaven代码3DNI [Oefner&Suck,19861)为例,对每个基序的分析和输出进行更详细的描述。 为此,该蛋白具有相当短的优势,并具有目前由PROMOTIF分析的大多数基序的实例
2.1 二结结构
蛋白二级结构的计算采用的 Kabsch and Sander (1983)定义的DSSP的算法。在标准的DSSP算法中,仅当其NH和CO基团形成适当的氢键时,该残基才包含在二级结构中,例如,对于β-sheets, 需要COi-1和NH<i+1>基团形成适当的氢键。这给出了与IUPAC规则6.2(1970)大致一致的赋值,该规则指出,要参与特定的二级结构,残的Φ和ψ 值应接近该二级结构的理想Vaues。
在这套程序中使用的稍作修改的算法符合IUPAC常规规则6.3,根据该规则,如果残基的NH或CO共基团参与了适当的氢键,则该残基被认为是β -sheet或α-螺旋的一部分。 在这种情况下,一个多余的残基在可能的情况下被添加到每条链和螺旋的末端,并被指定为小写字母“ h”和“ e”。 此规则是晶体学家中最常用的规则。 二级结构因此提供了用于其余分析的原始数据。
2.1.1 Turns
β-Turns
β-turns定义为四个连续的残基(用i,i + 1,i + 2,i + 3表示),其中残基i的Cα原子和i +3的Cα原子之间的距离小于7A,并且中心的两个残基不是螺旋的(刘易斯 等1973)。β-turns按照残基i + 1和i + 2的Φ和ψ 角分类(Venkatachalam,1968; Richardson,1981),每种转弯类型的理想角度如表1所示。 Φ和ψ 角度允许与这些理想值相差30°,其中一个角度的附加灵活性允许偏离40°。不符合上述任何标准的转弯归为IV型。

图1显示了由PROMOTIF为DNA转向DNASE I生成的彩色后记示意图的示例。该程序为蛋白质中鉴定出的每个β-turns生成Ramachandran图和示意图。 该蛋白质共有20个β-turns,其中8个在此显示

该程序还产生一个黑白表(表2A),提供有关每个β-turn的更多详细信息。 对于蛋白质中的β-turn和其他所有基序,还将创建提供大部分信息的机器可读平面文件(未显示)
γ-Turns
γ-turn定义为三个连续残基(i,i + 1,i + 2),在残基i和i + 2之间具有氢键,其中残基i + 1 的 Φ和ψ角在标准角的40°范围内。 标准角包括两类:经典和逆类( classic and invere)(表1)(Rose等,1985; Milner-White等,1988)。
跟β-turns一样,PROMOTIF输出包括一个彩色图,显示了蛋白质DNASE I中识别的每个γ-turn的Ramachandran图和示意图(图2)。更详细的信息在表中(表2B)和 平面文件

2.1.2 Disulphide bridges 二硫桥
对两个半胱氨酸残基鉴定了二硫键,它们的硫原子间隔小于3A。 理查森(Richardson 1981))根据二硫化物的内部χ角确定了几类,特别是x2,x3和x‘角度。 她发现大多数二硫化物可归类为左旋螺旋形或右旋钩形。 我们仅根据这些χ角的符号将二硫化物粗略地分类为四类(表3A)

PROMOTIF生成一张表(表3B),该表提供了蛋白质中发现的每个二硫键的详细信息。 DNASE I有两个二硫桥,其中一个被分类为右手hook,其中心χ角带有(-,+,-)符号,这是非常不寻常的。 第二个二硫化物甚至更不寻常,因为它的中心χ角具有(-,-,+)符号,因此它不属于上述任何类别
2.1.3 Helices
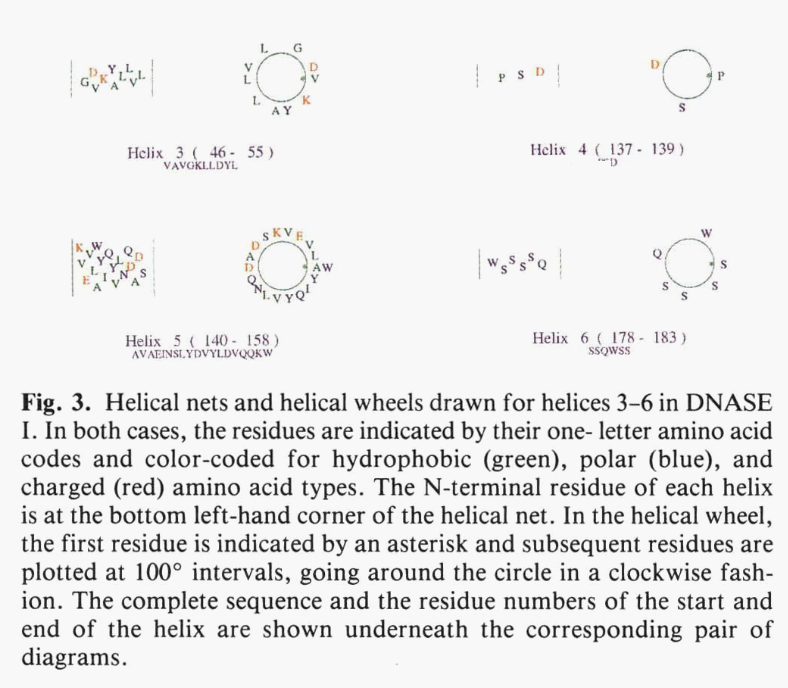
PROMOTIF生成一个表,该表提供有关由二级结构分配程序标识的每个螺旋的基本信息(表4)。 假设每匝有3.6个残基,则绘制每个螺旋的螺旋wheel和nets(图3显示了蛋白质中3-6螺旋的这些轮和网)。 最后,PROMOTIF提供了有关蛋白质中螺旋相互作用对的信息(表5)。 如果两个螺旋在另一个螺旋的一个或多个原子的4.5 A之内包含一个或多个原子,则视为“相互作用”(interacting)


2.1.4 β-Strands and β-sheet
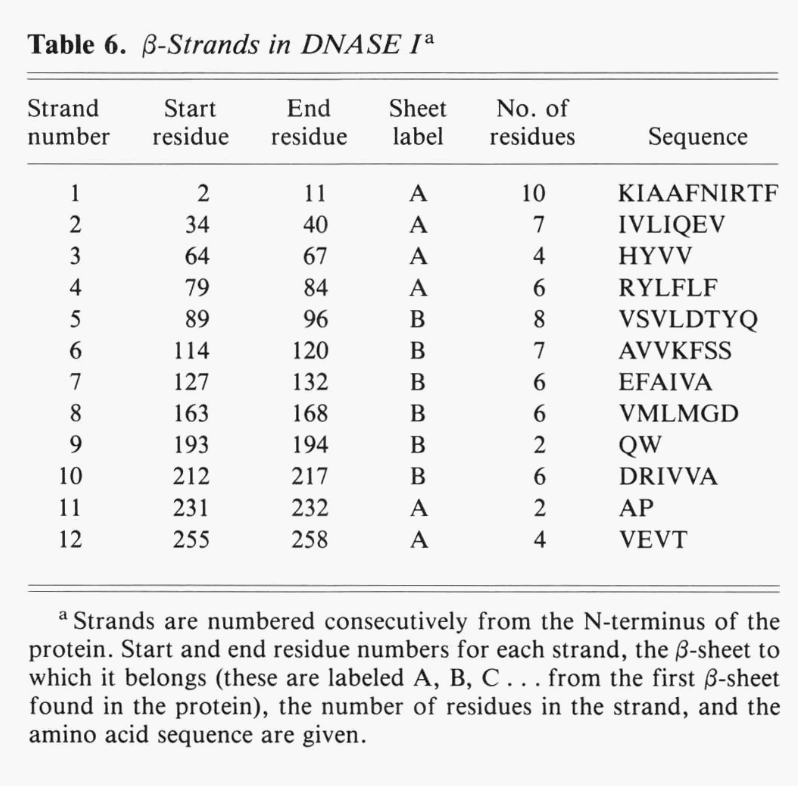
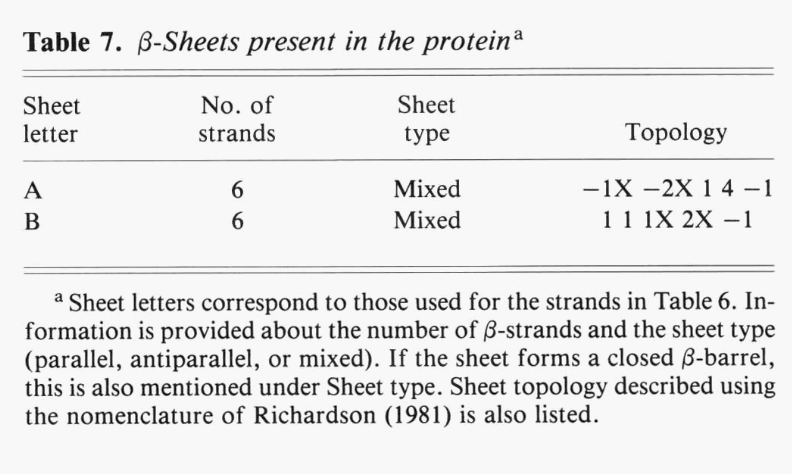
为每个β-Strands提供了更多基本数据(表6),而第二个表(表7)提供了有关每个β-sheet 的信息。 sheet的拓扑结构是使用Richardson(1981)的命名法给出的。 这为sheet中每对顺序的strand之间的连接分配了一个数字。 该数字表示连接在sheet上横穿的strand,以及在哪个方向上添加的“ X”用于交叉连接。 因此,β-发夹将具有“ +1”连接,β-α-β单元将具有“ + 1X”连接
2.1.5 β-Bulges
β-Bugles是β-shet中不规则的凸起区域,其中氢键的正常模式被破坏,例如,通过插入额外的残基。 PROMOTIF使用Chan等人(1993)最近描述的算法鉴定和分类蛋白质中的凸起。图4和表8显示了在DNASE I中发现的凸起。根据所涉及的两条链的相对方向,将凸起分为平行或反平行。在这些类别的每一个类别中,凸出部根据所涉及的残基数量和氢键结合方式进一步细分为经典,宽,弯曲,G1和特殊类型)( classic, wide, bent, G1, and special types)。因此,例如,隆起(bulge)可以描述为反平行经典(antiparallel classic)。经典的和宽的凸起物在其相邻链的一个β-sheet上涉及一个额外的残基。在反平行β-sheet中,经典的凸起出现在多余的残基位于两个间隔较窄的氢键对之间的情况下,而在凸起较大的情况下,多余的残基位于相距较远的氢键对之间。在Chan等人的文章中,可以找到parallel cIassic and wide bulges的相应氢键模式(1993)。弯曲凸起的发生频率要低得多,并且在两个链对上都有一个额外的残基。球状突起仅在反平行片中发生;在这些情况下,残基1 为αL构型,因此通常为甘氨酸。这通常发生在β-strand的末端。特殊的凸起涉及在一个链中最多插入三个残基,并且像弯曲的凸起一样,非常罕见
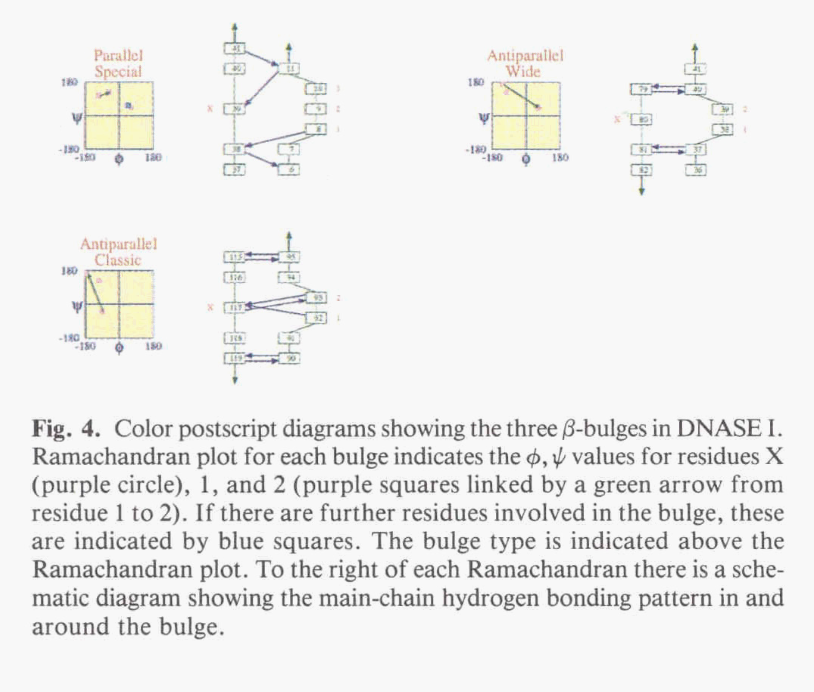
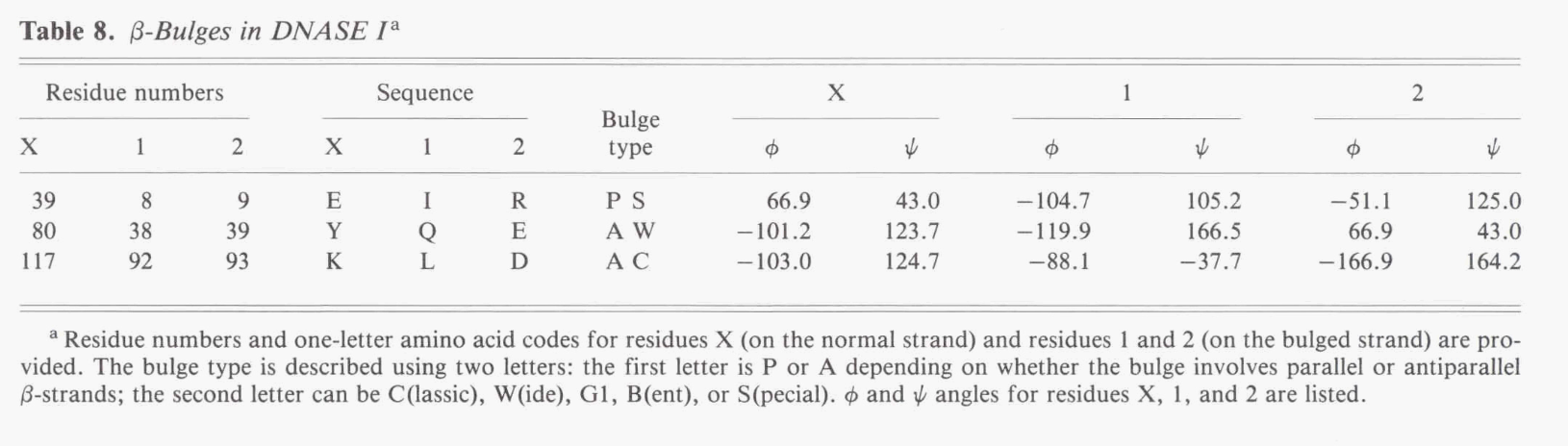
图4显示了DNASE I中每个凸出部分的Ramachandran图和氢键示意图。表8中显示了残基X,1和2的序列信息和详细的Φ和ψ值。有关残基3和4的详细信息, 以平面文件形式提供。
2.1.5 β-Hairpins
β-发夹由两个反向平行且氢键结合的β-strand组成。 Sibanda(1989年)将发夹分类,使用两个数字X:Y,表示使用两个不同IUPAC惯例(1970)定义的两条链之间的循环中的残基数。 如果末端链残基形成两个氢键,则X = Y。 如果未形成远端氢键,则环中的残基数取决于使用的链的IUPAC定义。实际上,如果未形成末端氢键,则Y = X + 2
对于较小的环路,发夹由β-turns(通常是I’和II’)的形成控制(Sibanda&Thornton,1985)。 3:5发夹由一个明确定义的构象主导,该构象可谓是I型转弯,后接G1凸起。 4:4发夹中最常见的类别包括I型P形转弯。 在出现这些特殊构型的地方,在主要分类后用适当的字母表示(例如:2:2I'; 3:5IG; 4:4I)。
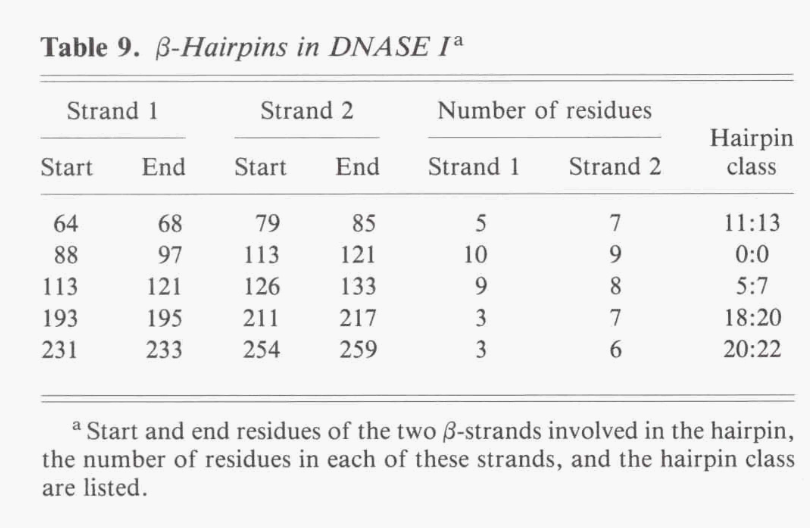
PROMOTIF绘制了蛋白质中每个发夹的示意图,显示了残基编号和发夹类别(图5)。 链和环中涉及的序列和氢键键合模式显示在右侧。 此信息以表格形式汇总(表9)

2.1.6 β-α-β Motifs and ψ-loop
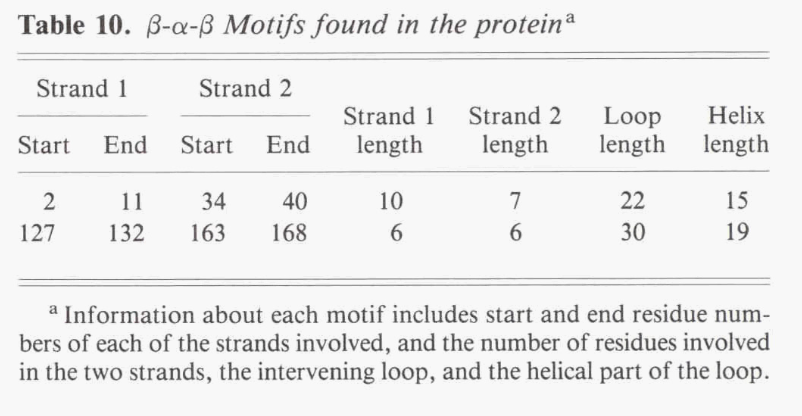
给出了蛋白质中 β-α-β 单元和ψ环的初步信息。 β-α-β单元由通过α-螺旋连接的两个平行的氢键合β-strand组成。 一个简单的表提供了有关这些位置的信息(表10)。
ψ-loop由通过“ +2”连接的两条反平行链组成,即,中间有一条链,氢键键合到两个环上(Tang等,1978)。 与β-α-β单位和β发夹相反,它们很少在蛋白质中发生(Hutchinson&Thornton,1990)。 同样,PROMOTIF提供有关任何ψ-loop位置的简单信息。 在DNASE I中未找到ψ-loop,但在找到它们的地方,由PROMOTIF生成的表的格式与为β-α-β基序生成的表类似(表10)。
2.1.7 Main-chain hydrogen bondingpatterns 主链氢键键合模式
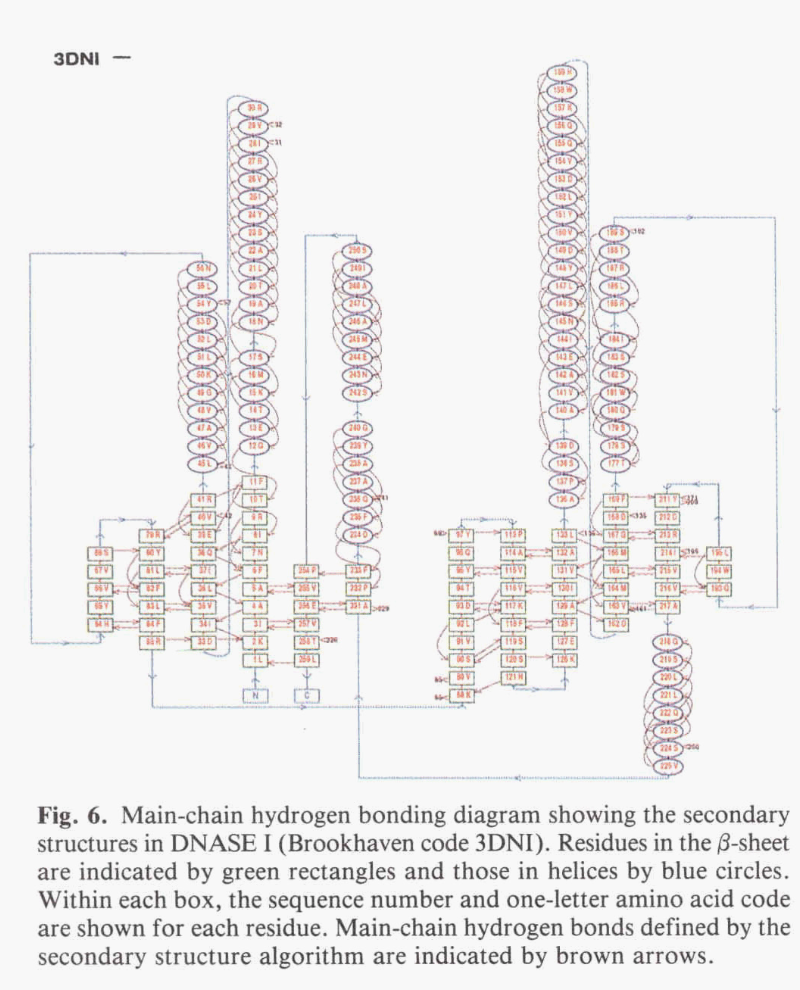
PROMOTIF将绘制一个由程序HERA(Hutchinson&Thornton,1990)绘制的彩色示意图,以说明蛋白质的β-sheets和螺旋中的主链氢键键合模式。 这应该有助于在整个蛋白质的背景下定位上述基序(图6)。
2.1.8 总结
上面每个主题的详细信息可能很冗长,并非总是必需的。 当只需要对蛋白质中的基序进行简要总结时,PROMOTIF也会生成一个摘要页面(图7)。 该页面的顶部概述了辅助结构信息,摘要页面的其余部分由微型表组成,这些微型表给出了结构中发现的每种主题类型的示例的位置和分类。

Φ和ψ
参考资料
- PROMOTIF–a Program to Identify and Analyze Structural Motifs in Proteins
- https://www.wwpdb.org/documentation/procedure
- https://pubmed.ncbi.nlm.nih.gov/8745398-promotif-a-program-to-identify-and-analyze-structural-motifs-in-proteins/?from_term=Promotif&from_pos=1
