【2.1.3】HSSP
HOMOLOGY DERIVED SECONDARY STRUCTURE OF PROTEINS
基于以下观察,可通过使用序列同源性显着增加已知蛋白质三维结构的数据库。
- 已知序列的数据库目前有12,000多种蛋白质,比已知结构的数据库大两个数量级。
- 目前预测蛋白质结构最有效的方法是通过同源性建立模型。
- 从序列相似性水平可以推断出结构同源性。
- 足以用于结构同源性的序列相似性阈值在很大程度上取决于比对的长度。
在这里,我们首先通过详尽调查已知结构蛋白之间的比对来量化序列相似性,结构相似性和比对长度之间的关系,并报告同源性阈值曲线作为比对长度的函数。然后,通过将所有根据阈值曲线被认为同源的序列与已知结构的每个蛋白质进行比对,我们产生了同源性蛋白质二级结构(HSSP,homology-derived secondary structure of proteins )的数据库。对于每个已知的蛋白质结构,派生数据库均包含比对的序列,二级结构,序列变异性和序列谱。隐含比对序列的三级结构,但未明确建模。该数据库有效地将已知蛋白质结构的数目增加了五到1800倍以上。该结果对于评估序列数据库搜索中的匹配结构的重要性,推导结构预测的偏好和模式,阐明结构残基的功能,以及通过同源性对三维细节建模。
数据库结构预测的局限性
给定一个新测序的基因,有两种主要方法可从氨基酸序列预测结构和功能。
- 同源性方法是最有效的方法,其基础是检测与已知结构的蛋白质或蛋白质家族特征性序列模式的显着延伸序列相似性。
- 统计方法不太成功,但更通用,并且基于单个残基,残基对,短寡肽或短序列模式的结构偏好值的推导。
两种方法都受到数据库大小的严重限制,在数据库中进行搜索或从中获得结构偏好。例如,为了在3个二级结构状态(螺旋,延伸链,环)的每一个中平均出现5种可能的8000(20的3次方)个氨基酸三联体,需要一个数据库,其中包含不同已知结构中约120,000个残基(8000 * 3 * 5)。所有四联体需要20倍以上数据量
关键技术问题
当序列相似性很高且长度延长时,将结构信息转移到潜在的同源蛋白上就很简单,但是当序列相似性很弱或局限于一个短区域时,很难评估序列相似性的结构重要性。请注意两个极端的例子。
1, 扩展的弱序列相似性但非常相似的结构:最佳重叠后的rus p21蛋白和延伸因子TU在链折叠的拓扑结构上相同,并且在总体结构上相似(rmsd只有2.4A ,Ca 166个残基中有138个的位置),但是这两种蛋白质的序列不同,残基少于20%。 2. 短强序列相似性但结构差异很大.枯草杆菌蛋白酶(2SBTI7和免疫球蛋白(3FAB))的八肽结构不同,具有4.7 A rmsD C(a)偏差,但在序列上有75%相同(图1)。
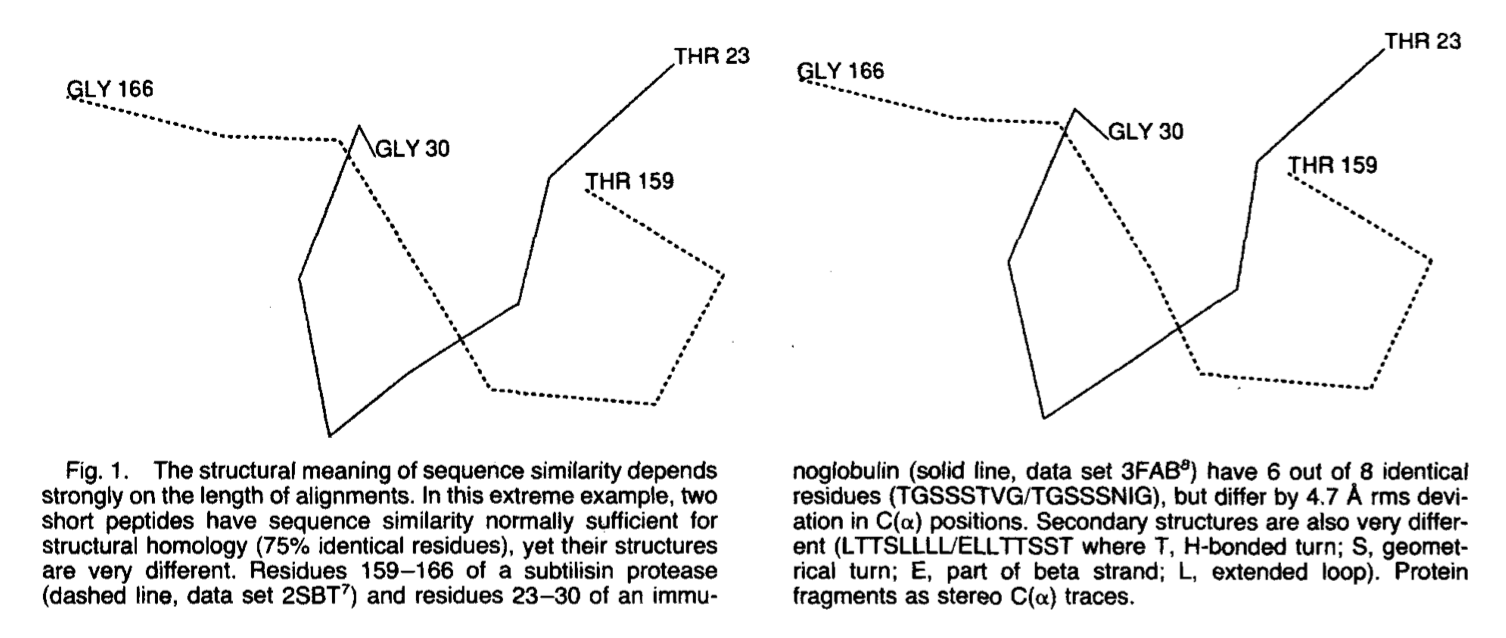
这些例子说明了两个关键问题之一(另一个问题是对序列相似性的精确测量):比对的长度越短,结构重要性所需的相似性水平越高。
同源性阈值与长度的关系
为了解决这个问题,我们需要校准序列相似性的结构重要性的长度依赖性。 根据经验,这可以通过从已知结构的数据库中得出序列相似性,结构相似性和比对长度之间关系的定量描述来完成。 长度依赖性同源性阈值的最终定义可以为可靠地推断球蛋白的可能结构降低至域和片段大小提供基础。 以前,Chothia和Lesk’已经量化了整个球形蛋白核心的序列相似性和三维结构之间的关系。
扩展已知结构的数据库
解决了长度依赖性问题后,我们可以通过穷举搜索相似序列片段来开始将序列数据库中的信息与结构数据库中的信息进行合并。 选择与已知结构的蛋白质具有显着序列相似性(同源性)的序列比对,即可得到一个同源性衍生的蛋白质结构数据库,比蛋白质数据库大几倍。

- 对于少于10个残基的比对,任何序列相似性值似乎都与任何结构相似度一致。
- 比80个残基更长的比对具有约25%相同残基的渐近阈值。
- 精确的数值取决于所用序列相似性的量度。 在这里,为简单起见,我们使用百分比相同的残基。



参考资料
- Database of homology-derived protein structures and the structural meaning of sequence alignment.Sander C and Schneider R, Proteins (1991) 9, 56-68.
