【4.1.1】蛋白质二级结构预测
蛋白质二级结构预测(PSSP)是蛋白质科学和计算生物学的基本任务,可用于了解蛋白质3维(3-D)结构,并进一步了解其生物学功能。在过去的十年中,已经提出了许多用于PSSP的方法。为了了解PSSP的最新进展,本文对这一领域的发展进行了调查。首先介绍了PSSP的背景和相关知识,包括基本概念,数据集,输入数据特征和预测准确性评估。然后,它回顾了PSSP的最新算法发展,主要侧重于最近的十年。最后,总结了该领域的相应趋势和挑战。这项调查得出的结论是,尽管已经提出了多种PSSP方法,但仍然存在一些进一步的改进或潜在的研究方向。我们希望提出的指南将帮助非专业人士和专家了解PSSP近年来的关键进展。
一、简介
蛋白质是任何活细胞中最重要的分子之一,也是所有活生物体的物质基础,这些生物参与了生命的所有过程,例如指导生化反应的催化,信号转导和传递或正确的表达遗传信息[1,2]。由于先进的测序技术的发展,大量的蛋白质序列数据可以以非常低的成本在数据库中获得;然而,人类只知道这些序列的结构总量约为0.2%,而公认的功能要少得多[3-6]。因此,从如此庞大的序列数据中了解蛋白质的结构和功能对生物学家和医学科学家来说仍然是一个巨大的挑战[1,7]。结果,通过使用先进的计算机技术来学习蛋白质的结构信息成为蛋白质科学和生物信息学的基本任务,并且可以用来了解蛋白质如何行使其生物学功能以及蛋白质与蛋白质分子之间的关系。
蛋白质的功能与其结构密切相关。因此,很明显,蛋白质组未知蛋白质结构的预测可能对发现其功能的任何尝试产生强烈的积极影响。通常,蛋白质结构层次分为四个不同的级别:一级,二级,三级和四级。蛋白质的三级结构和四级结构决定了蛋白质的3-D结构,并进一步决定了其功能特性。二级结构是一级和三级结构之间的桥梁,它是蛋白质分子的早期折叠阶段,是蛋白质3-D结构的基础。因此,蛋白质二级结构的研究是3-D结构研究中必不可少的第一步,也是最重要的一步,可以帮助理解蛋白质的功能与一级结构之间的关系[8]。除了学习蛋白质的3-D空间结构外,它还可以用于许多蛋白质科学领域[9],例如天然三级结构的预测[10],过渡态位置的预测[11],实际值溶剂可及性的预测[12],蛋白与蛋白的相互作用的预测[13],蛋白结构类别的预测[14-17],蛋白结构域的预测[18,19],蛋白的π-turns的预测[20]等。
由于基因组学和蛋白质组学的飞速发展;特别地,DNA测序技术和蛋白质测序技术导致蛋白质序列数据的大量积累。从序列数据中学习蛋白质的二级结构和三级结构是生物信息学的最大挑战之一。通常,在实验室中可以通过X射线晶体学和多维磁共振获得蛋白质结构,这些实验方法可以获得高精度的准确蛋白质结构信息。然而,这些方法的缺点是极其困难,成本高昂,费时,分子量有限。因此,实验方法显然不能应对快速增长的蛋白质序列数据的挑战[3-5,21]。由于蛋白质科学的飞速发展,蛋白质结构预测的计算方法是新兴的生物信息学交叉学科中最重要和最有效的技术之一,它具有简单,低成本和快速的特点,可以克服实验方法的缺点
PSSP计算方法的原理是,这些方法可以基于对已知蛋白质序列及其二级结构的分析来学习一些规则,以预测未知蛋白质序列的二级结构。 通常,它应该在PSSP方法中考虑以下问题:
- 如何通过训练和测试预测变量来构建或选择有效且可靠的基准数据集[23];
- 如何通过特征提取方法有效地从蛋白质序列中获得结构信息;
- 哪种分类算法可用于准确预测二级结构
Chothla和Levitt于1976年提出了第一种PSSP方法,然后将预测技术分成三代[8,24]。
- 第一代出现于80年代之前,通常利用分配给不同二级结构的单个残基的统计概率来计算。然而,这些方法的总体准确性低于60%,不能满足蛋白质3-D结构分析和预测的要求,代表性的是Chou-Fasman方法。
- 第二代大约出现于1980年至1992年; PSSP采用了先进的统计方法;滑动窗口考虑了邻近残基信息,此外,还考虑了蛋白质的其他特征信息(如物理化学信息)。这些方法可以在一定程度上提高预测的准确性,但总体准确性仍不到65%,典型的方法是GORIII。
- 第三代出现在1992年之后;这些方法通常使用多序列比对(MSA)profile文件(例如位置特定的评分矩阵)作为高级机器学习模型的输入,以预测蛋白质的二级结构。这些方法不仅考虑了氨基酸的组成和相互作用,而且考虑了更多其他特征,例如长距离相关性。代表性的方法是PHD和PSIPRED,这一代的总体精度约为76%–80%[25,26]
- 此后,我们发现PSSP技术在过去的十年中受到了越来越广泛的关注,并出现了许多新的性质和趋势,并且PSSP方法的发展如表1所示。由于新的进展,PSSP的准确性得到了进一步的提高。 混合模型和机器学习工具,例如神经网络的升级版,支持向量机,概率图形模型,模糊理论等[27,26]; 此外,基于优化的方法和集成学习方法也广泛应用于该领域。 此外,在分析了最近几年的大部分论文之后,这项工作认为这些PSSP方法的特点是它们同时考虑了进化信息,氨基酸组成和其他蛋白质的自然特性。
除了基于个体修改模型的PSSP方案之外,我们发现越来越多的学者认识到由于固有的弱点,个体模型的性能通常会受到一些限制。 不同的模型可以根据它们的不同特征提取相应的蛋白质特征。 因此,这些模型可能彼此互补,可以整合为一种混合模型,这是近年来的一种新趋势,例如多种模型和集成方法的结合。 经过对最近论文的统计分析,我们发现在过去十年中已经提出了许多出色的方法,并且预测精度已接近85%。 然而,为了更精确的3-D结构预测,需要对PSSP方法进行进一步的改进[3]。
为了概述PSSP的最新进展,本文对过去十年该领域的发展进行了调查。 在本文中,我们在第一部分中提供了PSSP的详细背景和介绍。 其次,我们对PSSP的最新进展进行了调查,以报告其研究现状,主要集中在最新趋势上。 最后,讨论了相应的趋势和局限性。 尽管提出了各种预测方法,但最后对PSSP的剩余挑战进行了简短讨论。 使用提出的指南将帮助非专业人员和专家了解PSSP近年来的关键进展。
在第2节中,我们将介绍PSSP的相关知识; 第3节提供了PSSP技术不同家族的分类学调查,以描述其发展。 第4节介绍了它的未来趋势和局限性。 第五部分总结了本文。
二、蛋白质二级结构预测的相关知识
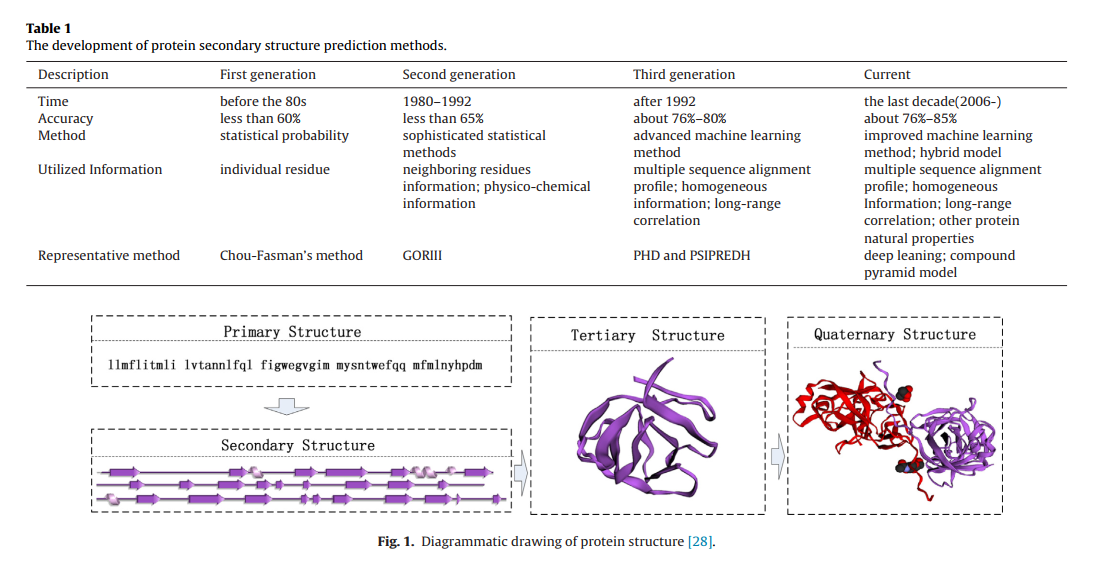
蛋白质结构层次可以分为四个级别:一级,二级,三级和四级,这四个蛋白质结构示意图如图1所示。一级结构仅是蛋白质多肽链的线性序列。二级结构是指多肽链的周期性结构片段。它是通过氢键的作用并在一维空间中沿着多肽链的方向生成的。三级结构是一条完整的多肽链,是由3-D空间中多个二级结构的进一步组合和折叠产生的,它可能已经代表了只有一条多肽链的那些蛋白质的主要生物学功能。四级结构是蛋白质复合物,由几条具有多个三级结构的多肽链组成,可以完全代表其生物学功能特征。
2.1. 蛋白质二级结构的描述
在这四个结构层次中,蛋白质的二级结构在蛋白质科学中起着重要的作用,它是多肽链的主要折叠,是蛋白质空间结构的基础。 通过多肽链内部氢键的影响,二级结构呈现出不同的状态。 最初,研究人员认为蛋白质中的氨基酸只有三个二级结构:螺旋(H),链(E)和卷曲(C) (Helix , Strand and Coil )。 这些二级结构可以反映氨基酸的局部空间排列:H是一种螺旋构型,通过每四个氨基酸之间的氢键得到增强; E是通过相互作用的氨基酸之间的氢键产生的平行的或反平行的链段结构; 对于那些不属于H或E类的氨基酸,C是默认类[84]。
此后,三态扩展为八态。 具体来说,蛋白质二级结构有几种分类方法,包括基于3-D空间中氢键的重复模式的蛋白质二级结构(DSSP)的定义,基于原子坐标的蛋白质二级结构分配(STRIDE)。 根据氢键和二面角的统计分布等分类策略。 这八类的不同分类方法将对预测结果产生很大影响[29],但DSSP是PSSP中最常用的方法,它将残基分为八个不同的二级结构:H(α-helix),G(310 -helix),I(π-helix),E(β-strand),B(isolated β-bridge),T(turn),S(bend)和C(others).
基于8-state结构的PSSP方法可以提供更详细的局部结构信息[75],但其预测将更加困难,直接导致其准确性低于3-state预测的12%[52]。 到目前为止,几乎所有方法都是基于三态的PSSP。 有五种流行的方法可以将8状态二级结构转换为3状态,包括方法1:H {H,G},E {E,B},C {S,T,I,C}; 方法2:H {H},E {E},C {G,S,T,B,I,C}; 方法3:H {H,G,I},E {E,B},C {S,T,C}; 方法4:H {H,G},E {E},C {S,T,B,I,C}; 方法5:H {H,G,I},E {E},C {S,T,B,C} [59],如表2所示。因此,应该指出,从3映射 态到8态显然是模糊的,因为它们的分类标准不同,其主要原因是在化学和生物学中对于这些二级结构没有非常明确的定义或边界。

2.2 数据集
在PSSP研究中,数据集的选择是至关重要的组成部分,因为它对PSSP模型的训练效果具有严重影响。 通常,数据集本身不代表任何特征,而仅代表分配给蛋白质序列的二级结构[67]。 在这些数据集中,经常使用CB513,CB396,RS126,EVA和PSIPRED
2.2.1 CB513和CB396
CB513数据集有513个序列,包含84107个残基,由Cuff和Barton提出[30]。 这是一个非同源且建立良好的基准数据集,所有513个蛋白质的序列相似性均小于25%,以确保在训练集中几乎没有同源性[78]。 它是最常用的独立数据集之一,包括CB396数据集和RS126的117个序列[61,31]。 CB396是一个非冗余数据集,具有来自CB513的396种蛋白质。 序列同一性百分比小于34%,平均序列长度为157个残基[32]
2.2.2 RS126
RS126具有126个蛋白质序列,包含26,846个残基,这也是Rost和Sandar开发的最常用的非同源数据集之一[33]。 平均序列同一性小于31%,平均序列长度为185个残基[61]。
2.2.3 EVA
EVA数据集由基于Web的服务器提供,名为自动蛋白质结构预测的评估。 它是蛋白质数据库(PDB)的最大序列唯一子集之一,并且在该子集中不存在比对的100个以上残基中具有超过33%相同残基的对。
GSW25和CASP数据集,包括其简化的数据集:CASP394和RCASP256(256种蛋白质)可用于盲法测试,但不适合模型开发[34,68,31]。 除上述数据集外,一些研究人员还使用PSIPRED,SCOP和SPINE [55,59,27]。 此外,还有一些其他的PSSP数据来源,例如核磁共振光谱[35],真空紫外圆二色谱光谱[36]和傅立叶变换红外光谱[37]等。
2.3 输入数据特征 Input data feature
输入数据特征是PSSP的另一个关键问题。 它是由数据集生成的,可以直接视为训练分类器的输入。 分类器的性能关键取决于对特征向量的明智选择,分类算法利用这些特征向量将特征空间划分为具有决策边界的不相交区域[102]。 这些特征可以有效地表示氨基酸序列的结构信息,并表示为一组数字,称为特征向量。 适当的输入数据特征将有效地提高PSSP的预测准确性[64],并且有许多针对不同预测方法的输入形式[41,119],可分为以下几类。
2.3.1 单序列
基于单序列的预测算法无法利用其他蛋白质的同源信息,因此其预测精度通常低于基于其他进化信息的方法。 但是,这对于蛋白质科学非常重要,因为在基因组测序项目中鉴定出的大多数蛋白质与任何已知蛋白质都没有可参考的序列相似性[87,128,129]。 此外,基于单序列的PSSP方法非常方便,并且在蛋白质科学的实际应用中简单易用。
2.3.2 多序列比对谱 Multiple sequence alignment profile
同源蛋白的多序列比对概况可以代表它们的结构比对和通常具有相似二级结构的比对残基[59]。 多序列比对谱可以通过许多方法生成,例如PSI-BLAST,PSI-Search,HMMER3,AMPS和CLUSTALW [31]。 多个序列比对可以产生特定于位置的概况,其提供有关结构的关键信息,并且可以用作PSSP方法的输入。 因为蛋白质的同源信息是预测未知蛋白质二级结构的非常可靠的支持。 BLOSUM62矩阵和PSSM矩阵是经常使用的多个序列比对配置文件,尤其是后者。 此外,一些研究人员还提出了其他方法来获得多个序列比对谱[38–41]。
2.3.2.1 Position specific scoring matrix
通常通过PSI-BLAST算法根据具有最高得分命中率的多个比对中每个位置的位置特异性得分的计算,来获得位置特异性得分矩阵(PSSM,Position specific scoring matrix)[31]。 每个氨基酸残基的特征信息估计为20要素特征向量,记录20列[77]。 高度保守和弱保守的位置分别由高分和接近零的分值表示。 PSSM矩阵可以有效地发现远距离相关的蛋白质序列和进化信息,因为它可以在不同位置反映不同的取代模式[42,71]。 PSSM可以极大地提高PSSP的精度,尤其是对于beta折叠而言,它是使用最广泛的方法
2.3.2.2 BLOSUM62矩阵。
BLOSUM62矩阵作为评分矩阵给出,由Henikoff和Henikoff提出。 它可以有效地测量两种蛋白质之间的差异,专门针对距离更远的蛋白质。 BLOSUM62矩阵用数值表示可能性的“对数奇数”得分,而可能性表示给定氨基酸对将互换的可能性。 在BLOSUM62矩阵中,具有相似化学性质的一对氨基酸更可能彼此取代,且给予正分。 相反,给定的具有非常不同的理化性质的氨基酸对则给出负分[41]。
2.3.3 理化性质
氨基酸的物理化学性质将对其蛋白质的二级结构产生严重影响,并可用于根据氨基酸的特性和残基之间的排列预测二级结构。为了分析蛋白质序列,根据其理化性质的相似性,可将20个氨基酸分为8种类型,即疏水性,亲水性,疏水性,亲水性和疏水性,极性,非极性;小大;已充电,未充电[43]。在蛋白质结构的研究中,可以使用8种代表性的理化性质来编码每个残基,并检查与蛋白质二级结构形成有关的相关信息。在这8个物理化学性质中,疏水,氢键和电荷性质对蛋白质的二级结构有很大影响,因此在PSSP中经常使用疏水和氢性质[31]。类似于BLOSUM62基质,疏水性基质也是基于物理化学性质的基质之一[41]
2.4 预测准确性评估
为了客观地评价PSSP方法的预测质量,大多数研究者广泛采用了许多评价指标。 预测质量评估方法通常用于测量PSSP算法的预测精度,并找到特定算法的最佳参数。 此外,还可以采用评估方法进行评估,并直观地表示PSSP方法的有效性。
最常用的评估方法是Q评分和分段重叠(SOV,segment overlap),它们是综合评估方法。 对蛋白质结构预测方法(CASP)的严格评估将SOV视为预测准确度的更合适方法[50,44]。 此外,还采用了其他一些方法来评估PSSP技术的性能,例如Matthews相关系数,平均绝对误差,平均绝对误差等。
2.4.1 Q score
PSSP方法的估计性能通常通过每个残基的三个状态准确度(Q3)或每个残基的八个状态准确度(Q8)评分功能进行评估,这是最简单,最受欢迎的度量方法,如(1),以及 Q分数可正确预测每个二级结构的残基百分比

2.4.2 细分重叠 Segment overlap
片段重叠(SOV)评分考虑了连续结构类型的片段,而不是简单计算正确残基的数量,这是基于观察到的片段与预测片段之间的平均重叠而不是平均每个残基的准确性,例如 (3)。 SOV可以容忍二级结构段末端的少量错误,但是会严重惩罚二级结构段中间区域的这些错误[45,52]

2.4.3 马修斯相关系数 Matthews correlation coefficient
马修斯相关系数(MCC)是一种更可靠的预测质量度量,在机器学习领域中经常用作度量二进制分类(两类)质量的相关系数[77,87]。 它同时考虑了过度预测和预测不足,并且通常被认为是一种平衡的衡量标准,即使类别的大小不同,它仍然可以使用。 它返回介于-1和+1之间的值,+ 1代表理想预测,0代表平均随机预测,-1代表逆预测。 为每种类型的二级结构定义了MCC,相应的公式表示为(5)

2.4.4 平均绝对误差
对于每种蛋白质,δ计算每个二级结构元素的平均绝对误差[75],如(6)所示。 δ是测量值与数据集平均值之间的绝对偏差的平均值,它实际上可以反映预测误

2.4.5 平均绝对误差 Mean absolute error
平均绝对误差(MAE)是观测值和预测值之间的绝对距离的平均值。 为了考虑二面角的周期性,通过[78]计算MAE:
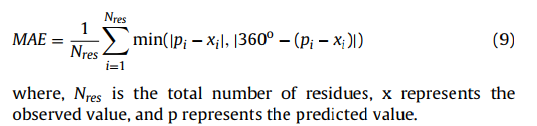
Besides, there are some less used indicators for the prediction quality analysis of PSSP, such as cross validation tests, self-consistency, standard error of prediction (SEP), k-state correlation coefficients, fuzzy Overlap (FOV), fuzzy correlation coefficient (Forr) and so on [45,75]
三、目前的方法
在本节中,我们将根据PSSP分类策略,介绍其最新发展,包括神经网络,支持向量机,概率图形模型,模糊理论和其他改进方法。 此外,在混合模型中,还特别引入了多种模型和集成方法的结合。
3.1 Neural networks
….
然而,由于梯度消失的问题,RNN在训练过程中通过梯度下降算法学习蛋白质的长期依赖性(longterm dependencies )也有一些局限性[57]。 而且,在正向链和反向链中的错误传播也受到指数衰减的影响,这使BRNN无法有效地学习蛋白质序列的远程信息[59]。
BRNNs无需使用窗口即可捕获远距离的相互作用,这可以避免基于滑动窗口(sliding window )的方法的缺点,并提取蛋白质中的远距离特征[60]。 两种改进的RNN版本都专注于模拟人类记忆形式,以更有效地捕获蛋白质的长期依赖性
…
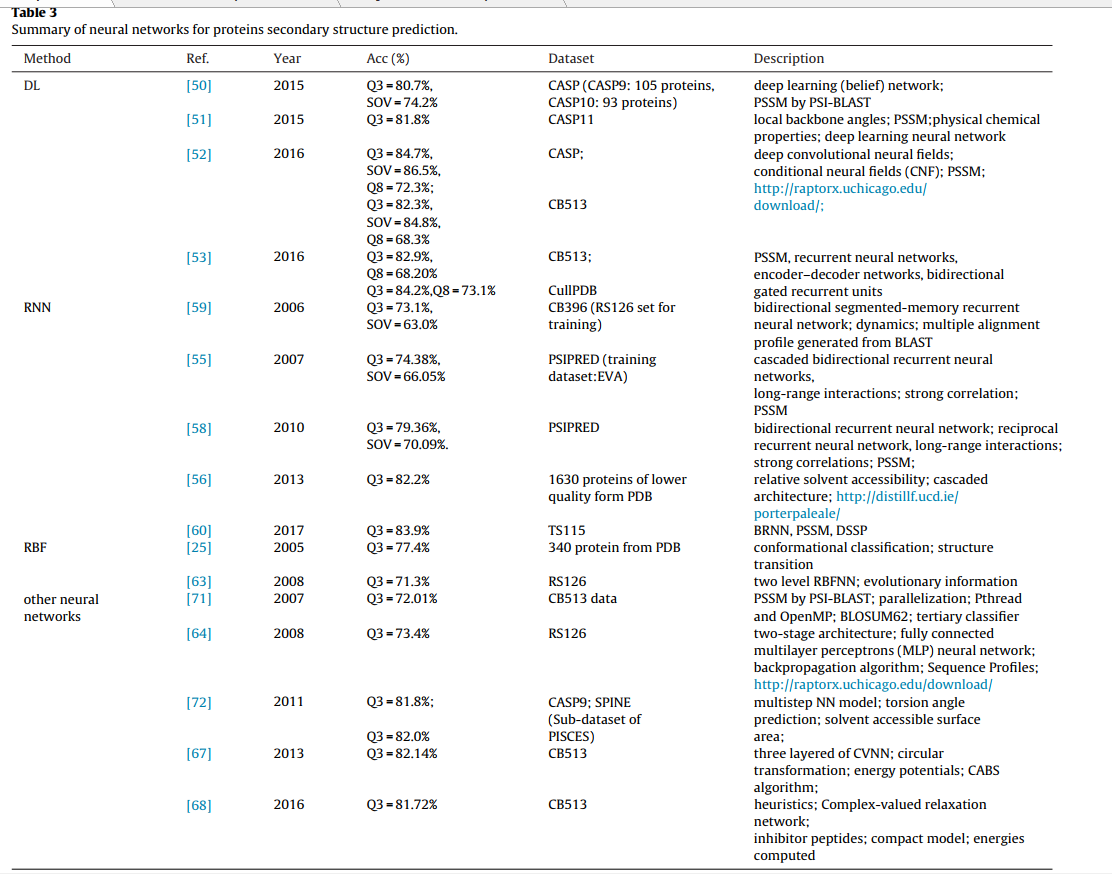
优点:
- NN不仅可以将多个序列比对图谱作为其输入载体,而且可以将蛋白质序列中相邻氨基酸的局部排列。
- NN在PSSP中的显着优势是其非线性映射能力,可以在训练过程中通过不同的构造规则来学习和存储很多输入和输出矢量之间的映射关系。
- 它可以完全近似蛋白质数据与二级结构之间的复杂非线性关系。 因此,它具有学习能力强,柔韧性好,容错能力强,自适应能力强的优点。 此外,它可以执行大规模并行处理并且易于设计。
缺点:
- 大多数NN无法有效捕获蛋白质中氨基酸的远程相互作用。
- 由于缺乏理论上的描述,几乎所有的神经网络都被认为是黑盒模型。
- 它没有解释如何达到预测结果以及为何做出决策的过程[119]; 此外,隐藏层和隐藏层节点的选择很困难,容易造成训练过度或训练不足。
- 它是如此地依赖于训练数据导致其对未经训练的新蛋白质的不良预测效果,并且它对输入数据编码方法也很敏感,从而导致其不同的预测效果。 通常,其培训过程通常也需要大量时间和样本
3.2. Support vector machines
…
为了提高PSSP的准确性,一些研究人员尝试将更多附加功能应用于SVM。 Kountouris等 认为蛋白质的骨架二面角与其二级结构高度相关,可以提供有关蛋白质局部3-D结构的关键信息,因此这项工作将二面角的独立结果和二级结构预测通过SVM组合在一起 提高预测准确性[78]。 此外,研究人员还提出了其他一些基于SVM的PSSP方法[41,79,80]。 此外,研究人员提出了其他一些基于SVM的PSSP方法,这些方法在第3.7节中进行了介绍[21,112,118,129]。
SVM由于其出色的模式识别能力而被广泛用于PSSP,非常适合PSSP任务的高维和非线性特征。 SVM具有多种内核功能,可以将非线性蛋白质数据映射到高维空间,并且此操作在低维空间中执行,以避免遭受“维数诅咒”的困扰。 因此,SVM适合PSSP的较大特征空间的特征; 它可以找到用于蛋白质结构分类的最佳分离超平面。 对于生物信息学,它还具有许多吸引人的功能,包括有效避免过度拟合,而过度拟合是监督学习技术的普遍缺陷。 需要指出的是,SVM最初是为二进制分类而设计的,即使有一些基于多类SVM的PSSP方法,但是如何更有效地对其进行改进以用于PSSP的多类分类仍是一个持续的研究问题[127]
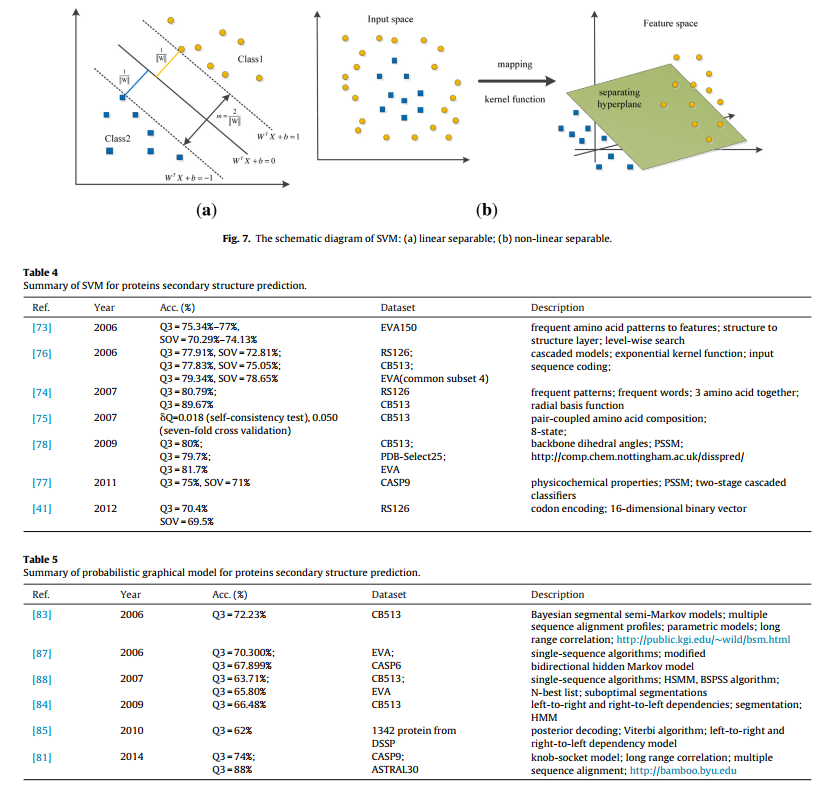
SVM由于其出色的模式识别能力而被广泛用于PSSP,非常适合PSSP任务的高维和非线性特征。 SVM具有多种内核功能,可以将非线性蛋白质数据映射到高维空间,并且此操作在低维空间中执行,以避免遭受“维数诅咒”的困扰。 因此,SVM适合PSSP的较大特征空间的特征; 它可以找到用于蛋白质结构分类的最佳分离超平面。 对于生物信息学,它还具有许多吸引人的功能,包括有效避免过度拟合,而过度拟合是监督学习技术的普遍缺陷。 需要指出的是,即使有一些基于多类SVM的PSSP方法,SVM最初也是为二进制分类而设计的,但是如何为PSSP的多类分类更有效地改进它仍然是一个持续的研究问题[127]。
3.3 概率图形模型 Probabilistic graphical model
概率图模型是概率论和图论的结合,它使用图来表示变量的联合概率分布。 贝叶斯网络和隐马尔可夫模型都是概率图模型的有向图,通常在PSSP中使用。 PSSP方法的概率图形模型摘要如表5所示。
3.3.1 贝叶斯网络
贝叶斯网络是有向无环图,它反映了不同变量之间的一系列概率依赖性关系,而没有考虑变量的时间因素。 当时间被认为是贝叶斯网络的一个附加因素时,它将变成动态贝叶斯网络(DBN),它可以通过其拓扑结构反映变量的时变和概率依赖性关系。
由于大多数概率方法对PSSP使用单个序列,因此,Li等人提出基于贝叶斯蛋白质包装的二级结构的贝叶斯模型用于PSSP,该模型考虑了残基对二级结构的包装影响,包括在空间上紧密但顺序较远的那些[81]。 并利用旋钮-插座模型为直接包含和预测线圈和匝的次级状态提供了构造。 这项研究表明,整合多个序列比对数据将提高PSSP的准确性。 此外,姚等还提出了一种基于DBN的方法; 它使用多元高斯分布来生成蛋白质序列的PSI-BLAST谱,其中考虑了PSSM条目之间的相关性
3.3.2 隐马尔可夫模型
作为一种特殊的贝叶斯网络,隐马尔可夫模型(HMM)是统计马尔可夫模型,是一种用于序列分析的实用工具,已在PSSP中成功实现。 在HMM中,状态不能直接观察,但是该模型的每个状态都通过相应的概率密度分布从一组观察中发出一个观察,如图8所示; 这些状态通常是蛋白质序列中的三个二级结构。 通常,HMM的观察结果被认为是氨基酸[82,84]
….
与NN和SVM相比,概率图形方法具有更明确的理论基础和计算过程,因此它们的机制更易于理解,例如相邻残基之间的特定相关结构[26]。 它可以用于通过基于结构片段的联合序列-结构概率分布来捕获蛋白质序列的远距离关系[46]。 然而,大多数概率图形方法被设计为在没有同源信息的情况下处理单个序列[87] [26],并且很难纳入不同蛋白质序列的进化信息,从而限制了PSSP的预测精度。
3.4 基于氨基酸组成统计(统计词典)的方法
众所周知,二级结构连续出现在蛋白质序列中,因此基于氨基酸组成统计(统计词典)的方法主要关注一定长度的连续氨基酸组成的统计特性。此外,这些氨基酸组合物的特征将以某种方式发现。然后根据发现的特征预测蛋白质的二级结构。在第3.2节中介绍了一些常见的连续氨基酸模式和基于SVM的方法。表6汇总了PSSP的统计词典模型。
Lin等人在PROSP [89]的基础上提出了一种改进的基于字典的PSSP方法,称为SymPred。通过合并SymPred和PSIPRED [90]也提出了一个名为SymPsiPred的元预测器。在他们的方法中,采用自然语言处理领域的同义词来捕获一组相似蛋白质中的局部序列相似性。对于PSSP,生成了蛋白质依赖性同义词字典[91],过程如图9所示。
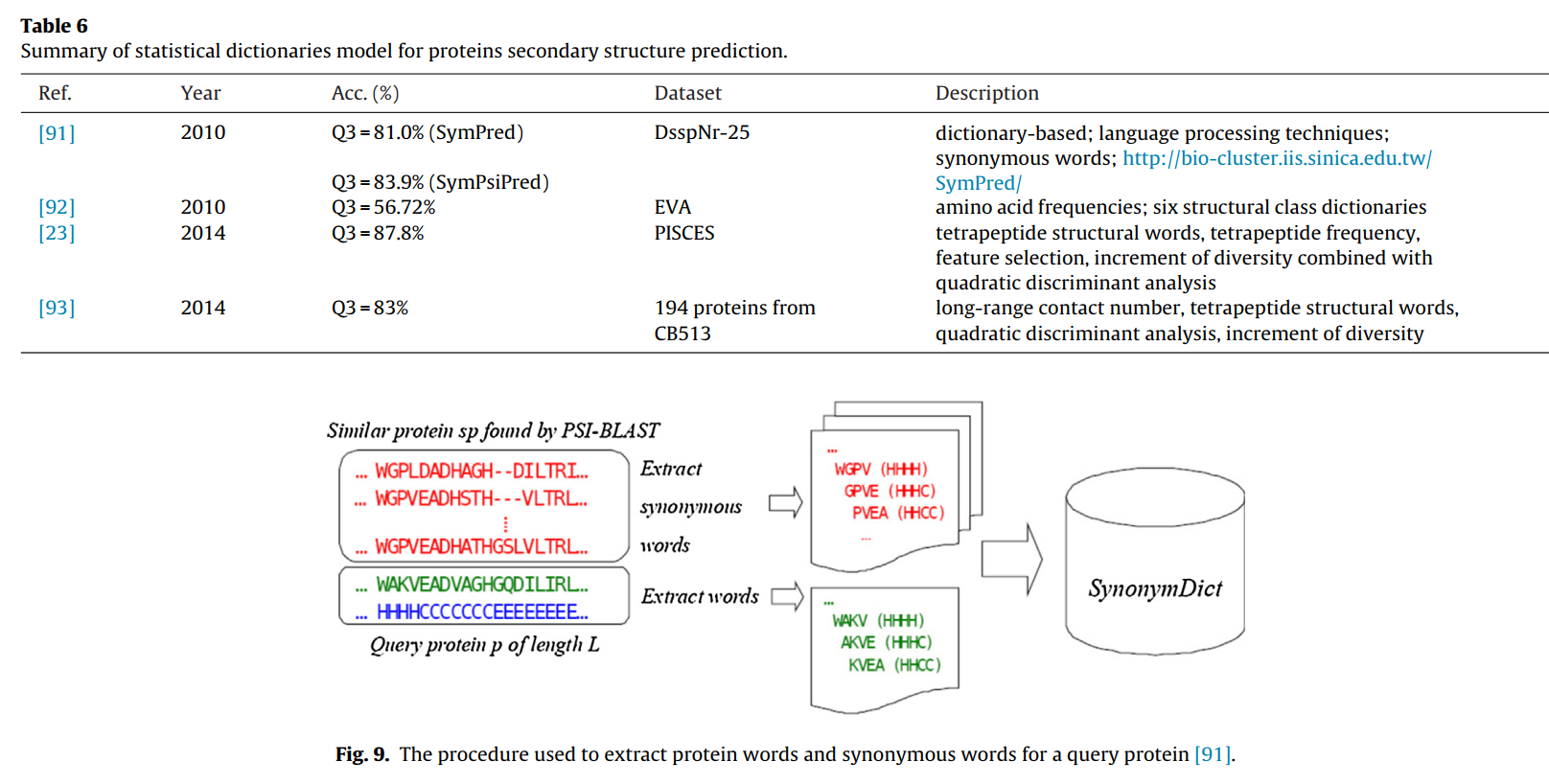
Popov等人根据氨基酸的频率词典,首先计算出每种氨基酸的总观察频率,并通过计算每个潜在二级结构的得分来确定每个位置上最可能的二级结构。 然后由六个结构分类词典为每个序列创建六个独立的预测; 最后,清除了明显错误或极低概率的元素[92]。 该方法的优点是它考虑了六个常见的氨基酸二级结构氨基酸序列读数。 而且该方法保持了较低的计算性能和与序列数据库的独立性
在Feng等人的研究中,四肽的频率特征是通过沿着蛋白质序列的四个残基的滑动窗口来计算的。 然后使用基于二项分布的方法对噪声和不必要的特征进行过滤,以找到具有高置信度的四肽结构词。 最后,通过多样性增量结合二次判别分析方法IDQD进行预测[23]。 在另一篇论文中,Feng根据四肽结构词的频率特征结合长距离接触数,提出了一种类似的PSSP方法,如果两个残基的分离超过蛋白质中的10个残基以上,则假定两个残基处于长距离接触 序列[93
上述工作证明基于频率词典的方法适用于PSSP,并且对于基于同源性的方法很少或根本不起作用的新型序列很有用。
- 这种方法的性能显然取决于小肽片段或连续氨基酸小片段的提取特性。因此,这些方法可以有效地发现蛋白质的局部特征。
- 但是,很难找到远程交互信息。固定长度组成统计方法将丢失非常短组成或长组成的信息,因为氨基酸的连续二级结构的长度是不确定的且具有多向性。
- 这些方法的关键问题是如何有效地设置阈值和提取局部二级结构信息。
3.5 Fuzzy logic
模糊逻辑系统可用于将人的推理编码到程序中以做出决策,该程序包含五个功能组件:模糊器,推理,去模糊器,模糊集和模糊规则(fuzzifier, inference, defuzzifier, fuzzy set, and fuzzy rule),如图10所示[94]。 通过使用模糊集的隶属函数,可以用[0,1] [95]范围内的标量来量化元素属性,如(28)和(29),它可以用来表示元素和集合之间具有不确定边界的隶属关系。 )[96] [97] [98]。 模糊推理应用满意的模糊规则将主要模糊化特征映射到其他次要模糊特征,并根据模糊规则通过确定的数学计算过程获得模糊结果。 模糊逻辑可广泛用于信息不完整或不精确的情况,因此它也可以有效地处理PSSP中的数据[45] [99] [100]。 表7总结了PSSP的模糊逻辑模型

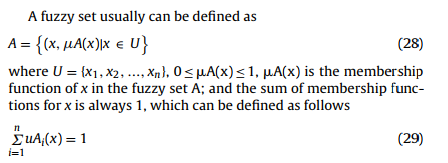
类型1模糊逻辑系统(类型1 FLS)利用隶属度来表示元素和集合之间隶属关系的不确定性。克里希纳吉等。为PSSP应用了Type-1 FLS组合遗传算法和神经网络。它利用模糊C均值聚类来减少属性输入的数量,这些属性包括蛋白质结构类别,溶剂可及性和理化性质。根据模糊规则库[94],使用模糊推理引擎将模糊预测转换为动作。类型2模糊逻辑系统(类型2 FLS)是类型1 FLS的扩展,它通过三维隶属函数描述类型1 FLS中成员函数的不确定性,并且可以更有效地处理不确定性。 Nguyen等。提出了一种用于PSSP的多输出间隔2型模糊逻辑系统(MOIT2FLS),这是该领域首次使用2型FLS。首先利用氨基酸的定量特性来表征20个氨基酸,这些氨基酸可以用作MOIT2FLS的输入.MOIT2FLS的三个聚类输出通过其自适应矢量量化(AVQ)方法分配给三个蛋白质二级结构。应用遗传算法优化了MOIT2FLS的参数[101]。
k最近邻(K-NN)方法是用于模式分类的最简单算法,很容易应用于并行计算。但是,它在PSSP中有一些限制。模糊理论与K-NN方法相结合,比对查询二级结构的确定预测提供更多的分类信息。 Kim提出了一种基于模糊k最近邻(FK-NN)方法的PSSP并行算法,该算法使用从PSI-BLAST获得的进化轮廓作为其输入特征向量[102]。邦杜古拉等人。也将FK-NN与用于PSSP的神经网络相结合,这将在3.7节中介绍[114]。之后,Ghosh等。通过使用三种低成本模式分类技术(如最小距离,K-NN和模糊k最近邻分类器)在PSSP中进行了尝试,而FK-NN在窗口大小为3时可以获得更好的性能[103]。
模糊逻辑具有处理不精确和模糊数据的能力,并且由于蛋白质结构预测的复杂性,该特性使其优于其他PSSP模型。它也可以估计氨基酸和二级结构类别之间存在关联的可能性,这是其他模型所缺乏的
3.6 其他方法
除了上述模型及其改进版本外,PSSP还使用了一些方法,例如RICO,Prote2S,K-nearest和LMNN。表8显示了其他用于PSSP的方法的摘要。
Lee等人基于覆盖规则的引入(RICO)方法[104]。通过使用基于规则的方法[105],提出了一种宽松的阈值RICO(RT-RICO)模型。 RICO利用粗糙集的一些概念,它是一种基于蛋白质数据集中实体划分的分类方案。 RT-RICO可以识别蛋白质序列中氨基酸之间的相关性,并为PSSP生成规则。但是这两种算法都存在不足,包括计算复杂度高和程序运行时间长。在另一位作者的论文中,通过并行化提出了一种略微改进的RT-RICO方法,以获得更快的运行速度[106]。
为了更有效地利用蛋白质数据库中的信息,并随着数据库的快速扩展提供不断提高的预测准确性,Chang等人。提出了一种使用核密度估计算法[107]的Prote2S预测器,该方法具有独特的优势,即在训练过程中时间复杂度低
。。。。
3.7 混合方法
。。。
四、未来趋势和局限性
PSSP是高度非线性和复杂的任务,可以看作特征提取和模式分类(features extraction and pattern classification)的综合问题。在本次调查中,我们发现PSSP方法的预测性能主要取决于机器学习技术的发展以及近十年来蛋白质的多种自然特性的考虑。前者主要依靠计算机科学的理论和技术进步,例如深度学习和数据挖掘,而后者主要依靠蛋白质数据集和生物学知识的进步。目前,大多数PSSP方法都基于通用的机器学习技术及其改进版本,因此也许应该通过关注蛋白质的自然特性为PSSP设计一些特殊的技术。 PSSP的进展将有助于3-D结构和功能预测的预测,并为生物学家和医学家提供有效的蛋白质结构信息。
4.1. 混合方法是一种趋势
混合方法是PSSP近年来的趋势,例如多种模型的组合,基于优化的混合方法和集成方法。先进的机器学习技术及其改进版本是PSSP性能不断提高的重要动力。但是,由于其独特的特性,单方法模型通常具有一些局限性。不同的模型可以根据其内置属性提取独特的蛋白质结构特征,这为通过将不同的个体方法集成到混合模型中来弥补其弱点提供了基础。
大多数混合方法对PSSP采用多步过程,通常由两个过程组成:蛋白质结构特征提取和二级结构预测。前者主要关注通过有效和合适的特征提取方法有效提取蛋白质数据的特征。后者采用了适当的方法来基于高级机器学习模型和分类器(集群)来预测二级结构
神经网络是混合方法中最流行的技术。它具有出色的非线性映射能力,可以在训练过程中通过不同的构造规则来建立输入和输出数据之间的关系。神经网络可以将蛋白质序列和多重序列比对概况作为输入,以考虑蛋白质序列信息和PSSP进化信息。但是,神经网络有一些缺点,例如由于缺乏理论上的描述,通常被用作黑匣子,并且考虑蛋白质的长度依赖性不是十分有效。幸运的是,有许多方法可以弥补神经网络的缺点,例如SVM,概率图形模型,KDD *,模糊k最近邻算法等,这些方法可用于增强对和PSSP方法的分类能力,提高预测精度。
除神经网络外,优化方法和集成学习方法也广泛用于PSSP的混合方法中。优化(Optimization)方法通常用于优化PSSP模型的参数或算法结构,以获得更好的预测结果。集成学习方法被广泛用于整合来自各种分类器的不同预测结果,以通过建立集成分类器模型来获得更好的预测性能。近年来,数据挖掘的概念也用于PSSP中,可以将其视为复杂的知识发现技术和机器学习模型的组合,以实现更好的预测效果。这些混合方法更加复杂并且需要更多的计算,并且可以被视为通过执行大量额外计算来补偿精度的一种方法,但是,由于它们的出色性能和光明的前途,它们仍然是一种趋势。
4.2 整合不同蛋白质的天然特性
蛋白质的自然特性,包括蛋白质序列信息,进化信息,局部依赖性,远距离信息,理化特性和生物学特性,应通过PSSP方法进行有效而合理的结合。 蛋白质特性的上述特征通常用作输入特征,这是PSSP的一个极为关键的组成部分。 PSSP分类器的性能关键取决于输入特征向量。 越来越多的研究人员认识到蛋白质的个别特性无法为进一步提高其预测准确性提供足够的信息。 结果,许多学者试图结合不同蛋白质特性的特征作为PSSP的输入,以达到更好的预测效果。
氨基酸的物理化学性质,氨基酸的局部空间排列以及蛋白质的长期相互依赖性(long-range interdependency)将严重影响蛋白质的二级结构,这反映了复杂的序列-结构关系和相互依赖性。这些蛋白质天然性质可用于编码每个残基,并检查与蛋白质二级结构形成有关的相关信息。进化信息由多个序列比对谱表示,其可以提供来自相似蛋白质序列的其他结构信息。除了这些常用的蛋白质特性外,一些研究人员还采用了其他蛋白质特性作为PSSP的其他功能,例如骨架二面角(扭转)角,溶剂可及表面积和基于Cα原子的二面体,它们也具有高度相关性具有蛋白质二级结构,并提供有关蛋白质局部3-D结构的重要信息。考虑到不同蛋白质特性的多重特征的方法被许多研究者采用,以进一步提高蛋白质二级结构的预测准确性。然而,如何从不同的蛋白质天然性质更有效地产生输入特征来表示其二级结构信息仍然是一个挑战,这也是一个值得将来研究的课题,因为它将为进一步提高PSSP性能提供许多可能性。
4.3 训练样本集和输入数据
选择有效的训练样本集是PSSP研究的重要基础,并且由于需要训练所有预测方法,因此对PSSP的性能具有严重影响。因此,训练样本的质量和数量将在预测模型中发挥重要作用。如何构建或选择有效和可靠的基准数据集以训练和测试预测变量是PSSP中的关键问题。特别是,某些PSSP方法对训练数据集非常敏感,应谨慎选择,这也会导致其局限性,例如鲁棒性差,可扩展性差和难以训练。结果,研究人员通过压缩或组合一些常用数据集为他们的PSSP方法提供了许多子数据集。但是,训练集必须足够大,并且按比例包括各种结构以实现更高的预测精度。
此外,一些机器学习算法由于其固有的属性而需要大量的样本,这会导致这些模型在训练数据有限的情况下表现不理想,例如深度学习和集成学习模型。甚至,研究人员提出了一些经过训练的小样本模型来克服此缺点,由于他们对此类复杂问题的知识有限,因此在实际PSSP应用中仍然存在一些对其性能的质疑。
通常,同源性或非同源性和序列同一性通常被用作选择训练样本的标准。 目前,大多数方法都采用多序列比对概况作为训练和预测的输入。 但是,数据集中同源或相似蛋白质序列不足的情况存在一些局限性。 因为考虑低同源性序列时,基于多序列比对的PSSP方法的准确性会大大降低。 基于单序列的预测算法无法利用其他同源蛋白质的进化信息,导致其预测精度较低; 此外,在基因组测序项目中鉴定出的大多数蛋白质与任何已知蛋白质均无参考序列相似性。 结果,新方法应该能够同时使用多个序列比对谱或蛋白质序列作为其训练样本或预测输入。
4.4 远距离相关 Long-range correlation
远程相关具有两个含义;第一个是在“不同蛋白质天然特性的整合”中提到的,它由数据格式表示,并且可以根据多序列比对情况用作PSSP的输入特征;第二是PSSP方法的能力,它是指预测方法可以有效地提取蛋白质中的长期依赖信息。如果PSSP可以考虑远程相关信息,则它可以实现更高的预测精度,因为这对于蛋白质中3-D结构的形成至关重要。结果,如何更有效地找到用于PSSP的蛋白质的长程相关信息(long-range correlation information)仍然是一个亟待解决的问题。
目前,预测精度高的方法通常具有强大的蛋白质数据知识发现能力。特别是,它们可以有效地提取远程依赖信息,例如基于深度学习和知识发现的方法。深度学习可以通过较低级别的功能(其众多的非线性操作元素)进行组合来发现更多抽象的表示形式。知识发现过程模型的基本机制是启发式协调器和维护协调器(heuristic coordinator and maintaining coordinator),可用于有效地发现蛋白质中的抽象特征。这两种方法对于PSSP都是潜在有希望的方法
4.5 鲁棒性和适应性 Robustness and adaptability
PSSP是一个高度动态且复杂的问题,包括特征提取和模式分类,这对PSPP方法的鲁棒性和适应性提出了巨大挑战。甚至许多基于机器学习技术的方法都可以在PSSP中获得良好的预测结果,但是很少有方法能够获得超过85%的预测精度。因此,提高PSSP的鲁棒性和适应性是提高其预测精度的关键。近年来,研究人员主要依靠混合方法来提高二级结构的预测性能,但是由于它们依赖于各个机器学习模型,因此存在一些限制。
近年来,随着先进测序技术和蛋白质科学的发展,数据库中蛋白质数据的数量不断增长。因此蛋白质序列数据的动态性将越来越高。目前,大多数PSSP方法无法应对蛋白质数据的增量特征。特别是,使用最广泛的基于神经网络的PSSP方法在新增数据中的适应性较差。更不幸的是,只有少数研究人员提到他们的方法可以应付PSSP的增量研究任务。因此,应积极追求具有更好的鲁棒性和适应性的PSSP方法,以实现更好的预测性能。 PSSP的强大鲁棒性和适应性将引领其在3D结构预测以及蛋白质科学甚至生物信息学和生物学的许多其他领域中的应用。
五、结论
PSSP是计算生物学和蛋白质科学领域的重要研究领域,是学习蛋白质3-D结构和生物学功能的一项基本任务。但是,由于最近十年来不断增长的需求,已经提出了许多新的PSSP方法。但是,它仍然不能满足蛋白质3-D结构和功能预测的需要,并不能为生物学家和医学家提供足够的蛋白质结构信息。本文提供了有关该领域的调查,以了解最新进展并尝试促进PSSP的发展。在本次调查中,我们首先提供PSSP的介绍和相关知识;然后,报道了PSSP的最新算法进展,以证明其研究现状。最后,讨论了相应的趋势和挑战。我们认为,尽管已经提出了许多预测方法,但是仍然存在的挑战和需求将进一步促进这种技术的发展。这项工作认为有一些可能性可以改善PSSP的性能:(i)混合方法是PSSP的趋势,因为它们可以整合单一模型的不同优势; (ii)不同蛋白质天然特性的整合将提供更多的蛋白质生物学信息; (iii)精心选择的训练集以及蛋白质中长距离相关性的考虑能够构建有效的PSSP模型; (IV)利用多序列比对谱可以提供来自其他已知蛋白质的更多生物学进化信息; (V)使用强大的机器学习技术将增强PSSP方法(如深度学习)的可靠性。因此,我们认为高效的PSSP方法可以为生物学家和医学家提供有关蛋白质结构的更准确信息。
参考资料
-
- Protein secondary structure prediction: A survey of the state of the art
