【4.1.2】药效团(Pharmacophore、QSAR)
一、基本概念
- 药物分子与受体靶点发生作用时,分子中的基团对于活性的影响不同
- 1909年,Paul Ehrlich提出,指载有活性必须的特征原子的分子框架。
- 1977年,Paul Ehrlich提出,指分子中的一组能够识别受体,并能形成分子生物活性的结构特征。
- 泛指药物活性分子中对活性起着重要作用的“药效特征元素”及其空间排列形式、包括了结合特征、结构和特性约束的信息,以作为数据库检索的提问方式。
在药物分子和靶点发生相互作用时药物分子为了能和靶点产生好的几何匹配和能量匹配会采用特定的构象模式即活性构象。而且对于一个药物分子分子中的不同基团对其活性影响是不同的有些基团的改变对分子活性的影响甚小而另外一些基团的变化则对分子与靶点的结合起着非常重要的影响。于是就需要引入一个药效团(Pharmacophore)的概念。
药效团是分子特征的抽象描述,其是生物大分子对配体的分子识别所必需的。 IUPAC(International Union of Pure and Applied Chemistry)将药效团定义为“空间和电子特征的集合,这是确保与特定生物靶标的最佳超分子相互作用以及触发(或阻断)其生物反应所必需的”。[1] 药效团模型解释了结构多样的配体如何与共同的受体位点结合。 此外,药效团模型可用于通过从头设计或虚拟筛选,鉴定将结合相同受体的新配体。
例子
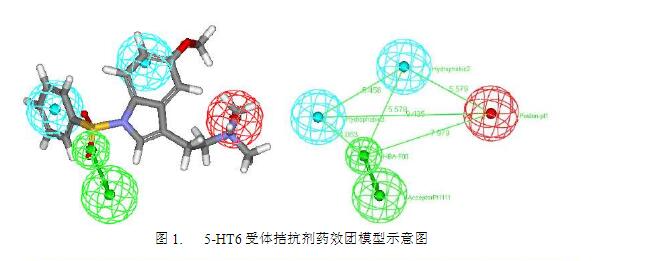
图1展示了一个经典的5-HT6受体拮抗剂药效团模型。该药效团模型由4个药效特征组成其中两个为疏水团(蓝色球所示),一个为正电基团(红色球),另一个为氢键受体基团(绿色球,含方向)。各个药效特征之间存在几何约束,相互之间的距离以及角度需满足一定限制条件。任何药物分子如果能够满足这一个药效团,就具备了与5-HT6受体结合的必要条件。
- 药效团(Pharmacophore):是一组在分子间相互作用中起关键作用、 并引起生物响应的立体和电子结构特征(steric and electronic features)。
- 药效团模型(Pharmacophore model):是指药物活性分子中对活性起 着重要作用的“药效特征元素” 及其空间排列形式。
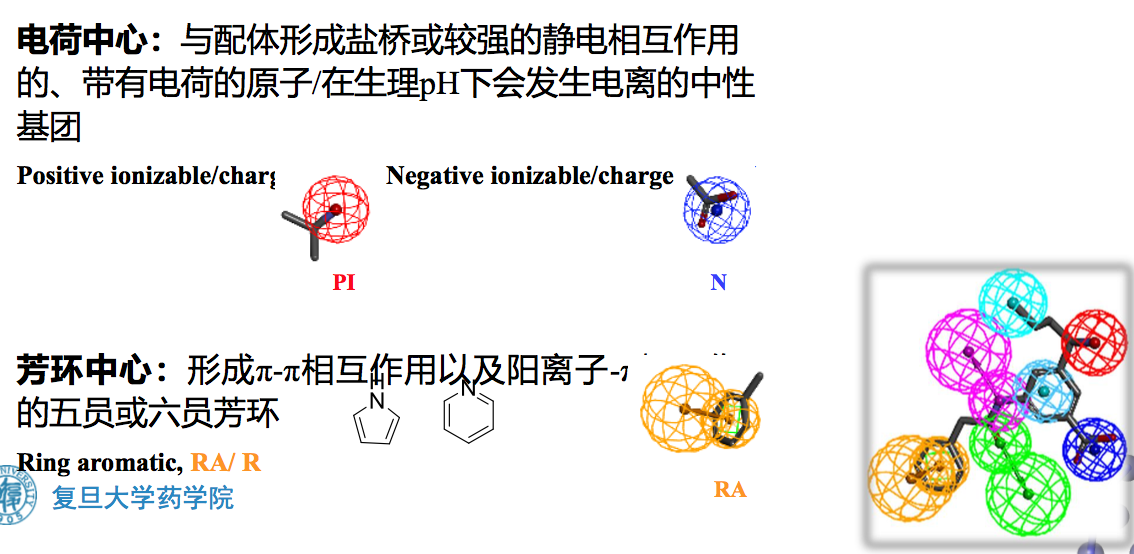
- 药效特征元素(Chemical Features) :包括氢键供体、氢键受体、正电 中心、负电中心、疏水中心、芳环中心和自定义的其他特征。
二、药效特征元素定义
在早期的药效团模型中,药效团模型的提问结构中一般只包括一些具体的原子或原子团,比如氮原子、羧基、苯环等。但是从药物分子与受体相互作用的角度看,药物分子中某个位置上并不一定必须包含某种特定原子或者原子团才能产生必需的特征相互作用。例如:药物分子上某个原子能作为氢键受体和蛋白产生氢键相互作用,那么这个位置上的原子既可以是O,也可以是N和S。在这种情况下,仅仅在药效团模型中定义具体的原子或原子团显然是不够的,需要引入更一般化的基于功能的药效特征元素。这些药效特征元素特指一般化的化学功能结构,比如氢键受体、氢键给体、疏水中心、芳环中心、正负电荷中心、排斥体积等。下面将对这些药效特征进行简要的介绍。
- 氢键受体
- 氢键給体
- 疏水中心:制药和不带电原子或电负性中心相连的一组连续的碳原子都可以形成疏水中心
- 电荷中心:可能能够与受体形成盐桥或较强的静电相互作用
- 芳环中心:形成π-π相互作用
这些药效点可以位于配体本身上,或者可以是假定位于受体中的投射点。
这些特征需要匹配具有相似性质的不同化学基团,以便鉴定新的配体。 配体 - 受体相互作用通常是“极性阳性”,“极性阴性”或“疏水性”。 明确定义的药效团模型包括疏水体积和氢键载体。
药效特征元素(不同颜色的球)

(一) 氢键受体
氢键相互作用是配体与受体之间相互识别非常重要的相互作用。因此氢键特征在药效团模型中占有重要地位。氢键特征可以分为两类:氢键给体和氢键受体。
广义来讲,任何带有孤对电子的原子。如氮、氧、氟、硫等,都可以作为氢键受体。但过于宽泛的定义往往会导致过多的命中结构,从而降低搜索的选择性。因此在一般的药效团模型方法中(Discovery Studio, Catalyst)仅仅只考虑药物分子中最常见的氢键受体形式,包括:
A. sp或sp2杂化的氧原子 B. 与碳原子以双键形式相连的硫原子 C. 与碳原子以双键或三键相连的氮原子
(二) 氢键给体
氢键给体主要包括氢原子以及与之相连的氧原子和氮原子,一般有
A.非酸性羟基 B.氨基 C.次氨基,但不包括三氟甲基磺酰胺和四唑中的次氨基。
需要特别指出的是,由于配体与受体之间的氢键相互作用一般具有明确的方向性,因此对于一个氢键给体或受体的描述,仅仅靠一个点是不够的。在药效团模型软件中(Discovery Studio, Catalyst),一般都采用两个点来描述氢键特征.一个点表示氢键特征中重原子的空间位置,而另一个点表示氢键给体或受体的矢量方向。对于氢键给体矢量方向为重原子和与之相连的氢原子的成键方向。对于氢键受体,矢量方向一般为重原子和其上孤对电子连线的方向。如下图所示,为Discovery Studio软件包中氢键给体(紫色)和氢键受体(绿色)特征的示意图。在比较一个药效团和测试分子中的氢键特征时,不仅要比较氢键特征的位置,还需要比较氢键特征的矢量方向。

(三) 疏水中心
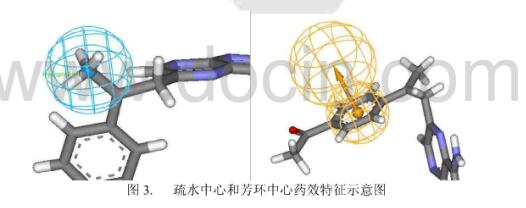
疏水相互作用是配体与受体相互识别的重要作用方式。配体与受体上的疏水基团总是倾向于形成紧密的疏水堆积作用,形成疏水性内核。疏水相互作用本质上包含了熵效应和范德华相互作用两个部分。疏水基团一般由非极性原子组成,有疏水相互的片段很多,如甲基、乙基、苯环等。
与氢键给体、氢键受体特征不同,疏水中心无需用矢量表示,只需要用一个点表示配体与受体形成疏水相互作用的部位就可以了,见下图。
(四) 芳环中心
芳环可以参与药物分子和蛋白受体之间∏电子离域系统的∏-∏相互作用。芳环中心主要包括五元和六元芳环,如噻吩、苯环等。在药效团模型方法中,芳环需要由两个参量来定义:一个参量是芳环的空间位置,即芳环中所有原子的几何中心;另一个参量是芳环平面矢量方向,一般用垂直于芳环平面的矢量来描述,见下图。
(五) 电荷中心
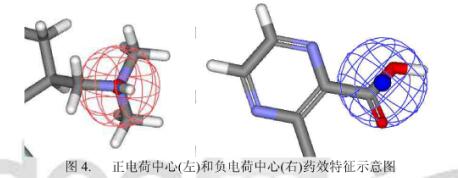
配体上的电荷中心是指配体上的带电基团,由于具有较多的部分电荷。这些基团往往可以和受体形成盐桥或较强的静电吸引作用。电荷中心既可以是带有电荷的原子,也可以是在生理PH下会发生电离的中性基团。比如:在生理PH下,脂肪胺会质子化形成正电荷中心,而羧基会去质子化形成负电荷中心。此外,∏电子离域系统,如羧酸盐、胍基、脒基等也可能形成电荷中心。电荷中心可以分为两类:正电荷中心和负电荷中心。
正电荷中心包括
A.带正电荷的原子 B.伯、仲、叔脂肪胺中的氮原子 C.氮-氮双取代的脒基中的亚氨氮原子或四氮取代的胍基中的亚氨氮原 D.至少含有一个未取代氢原子的脒基中的氮原子中心或至少含有一个未取代氢原子的胍基中的氮原子中心。
负电荷中心包括:
A.带负电的原子 B.三氟甲基磺酰胺中的氮原子 C.羧酸、亚磺酸或磷酸中羟基氧和氧代氧的原子中心 D.磷酸二酯和磷酸酯中羟基和氧代氧的原子中心 E.硫酸和磺酸中羟基氧和两个氧代氧的原子中心 F.磷酸单酯和磷酸中氧代氧和两个羟基氧的原子中心 G.四唑中的氨基氮原子
在药效团模型方法中,正负电荷中心都是由一个电荷中心点表示其所在位置,没有方向性,见下图。
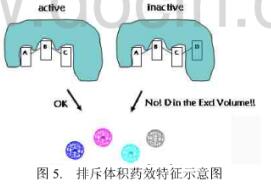
(六) 排斥体积
在药效团模型中,排斥体积也是一种重要的组成成分。与其余药效特征不同,排斥体积并不是对应于配体上特殊的原子或者基团,而是基于配体分子的空间特征。在配体和受体相互作用时,在配体的某些取代位置上存在某些原子或原子团可能会和受体产生不利的原子碰撞。这些位置上的原子或原子团占有的位置就构成排斥体积。在排斥体积中存在原子或原子团会大大降低化合物的活性,见下图。
(七) 药效特征的几何约束
一个完整的药效团模型中除了必须包含药效特征元素(如氢键给体、疏水中心、正电中心等)之外,还需要包括药效特征元素之间的空间约束。这些约束是指各特征元素的位置约束,各特征元素之间的距离、角度、取向等。
在常用的药效团模型方法中(如Discovery Studio, Catalyst),药效特征元素可以抽象为点(比如疏水中心、电荷中心)、线(比如氢键)、面(比如芳环平面)的形式出现。而这些特征元素或它们之间几何关系的约束可以采用多种形式来实现,如:位置约束可以是点的空间活动范围;距离限制可以是点点间的距离,或点到线的距离。角度限制可以是三点的角度,直线与平面的角度,或者是平面与平面的角度等(参考图1)。其中距离限制是最为常见的约束形式。但是仅仅限制药效特征元素之间的相对距离,无法反映化合物的立体选择性,不能表达手性的要求。因为一个构象中原子的相对关系和它的镜像是完全相同的,但是它们的活性可能完全不同。比如S,S型的captopril有ACE抑制活性(一种血管紧张素转化酶抑制剂),而R,R型却没有活性。在这种情况下,就需要添加更为严格的角度、二面角以及位置约束,才能得到特异性比较高的药效团模型。
药效特征元素的要素
- 药效特征元素(Chemical Features)
- 每个化学功能用不同颜色的球表示
- 位置和方向(Location and Orientation)
- 以绝对坐标定义,因此可区分对映体
- 距离和位置许可偏差(Tolerance)
- 球的大小代表位置精准度
- 排除体积(Excluded Volumes)
- 空间约束形式,可区分活性和非活性分子结构差别或限制配体和受体产生不利碰撞区域
三、应用
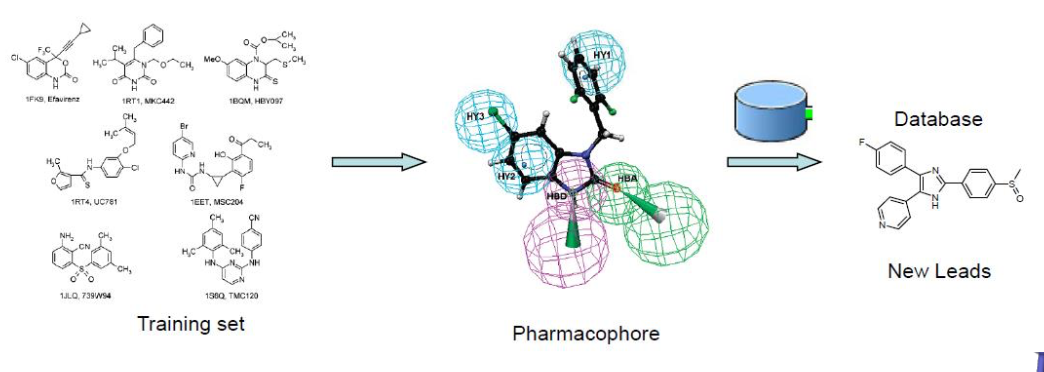
利用分子的三维结构信息进行药物分子设计已经成为药物化学领域一项常规的技术。依据 所依赖结构的不同(受体的三维结构或者配体的三维结构)药物设计的方法又可以分为两种 基于(受体)结构的药物设计方法和基于配体结构的药物设计方法。当受体(蛋白质、酶或DNA) 的三维结构已知时可以采用基于结构的药物设计方法采用分子对接或从头设计技术通过 研究配体与受体之间的相互作用信息进行药物设计。而当受体结构未知时则可以通过已知活 性的配体分子的三维结构建立恰当的构效关系模型来指导进行药物分子结构的优化和改造 这种方法叫做基于配体结构的药物设计方法。药效团模型方法就是一种最为杰出的基于配体结 构的药物分子设计方法。
在现代计算化学中,药效团用于定义具有相同生物活性的一种或多种分子的基本特征。 然后可以搜索不同化学化合物的数据库以寻找具有以相同相对取向排列的相同特征的更多分子。 Pharmacophores也被用作开发3D-QSAR模型的起点。 这些工具和“特权结构”的相关概念,“定义为能够通过明智的结构修饰为多于一种类型的受体或酶靶标提供有用配体的分子框架”,[3]有助于药物发现。
作为基于配体结构的药物设计中的最主要的两种方法,定量构效关系方法和药效团模型法。虽然都是以配体小分子的结构作为起点,但二者之间存在明显的不同。定量构效关系方法一般用于研究一些列骨架相同的同系列化合物,所得到的定量构效关系模型只适用于指导这一系列化合物的改造和活性预测。而药效团特征是对配体小分子活性特征的抽象与简化。也就是说制药小分子拥有药效团特征,就可能具备某种生物活性,而这些活性配体分子的结构未必需要相同,因此药效团模型方法可以用来寻找结构全新的先导化合物。
药效团模型方法包括两个层面的内容:
- 药效团模型的构建。从一系列活性小分子出发得到合适的药效团模型
- 数据库搜索
药效团模型的构建是指从一系列活性小分子出发得到合适的药效团模型。而如果想通过药效团模型来找到新的先导化合物,就需要采用基于药效团模型的数据库搜索。通过数据库搜索可以寻找包含特定药效团特征的化合物,这些具有特定药效团特征的化合物可能具有相应的生物活性。药效团模型方法作为一种发现先导化合物的有效方法,在药物研发领域已经得到了广泛的应用。近年来文献也报道大量通过基于药效团的数据库搜索方法找到先导化合物的成功实例。可以预见随着小分子三维结构数据库信息量的迅速增加,以及计算机技术的快速发展药效团模型方法在药物设计中会受到越来越多的关注。
四、药效团构建
药效团构建是从一组结构多样性的小分子出发,寻找其共同的药效 团特征,从而发现影响生物活性的关键因素,为药物设计提供有价 值的参考。
4.1 实验内容
采用一系列选择性雌激素受体调节剂(SERMs)构建一个3D QSAR药效团模型,用于预测新型小分子的活性。
所需功能和模块:
- Discovery Studio Client
- DS Catalyst Hypothesis
- DS Catalyst Conformation
- DS Catalyst Score
所需数据文件:
- serm_ligands.sd
- predict_serm_ligands.sd
- test_serm_ligands.sd
4.2 实验步骤

对训练集分子的要求
- 分子结构具有多样性
- 分子的活性值至少跨越4个数量级
- 每个活性水平的化合物数量至少为3个
- 化合物数目在16-31个
- 必须包含 Activ 和 Uncert 性质

Activ:化合物的实验活性值,如IC50,Ki。活性测试方法尽量一致, 或者是同一课题组测得的活性值;
Uncert:活性值的不确定度。该值必须大于1.0,默认为3.0。若 Activ为0.01,Uncert为3.0,则化合物的活性范围在 0.01/3=0.0033 和 0.01*3=0.03 之间。
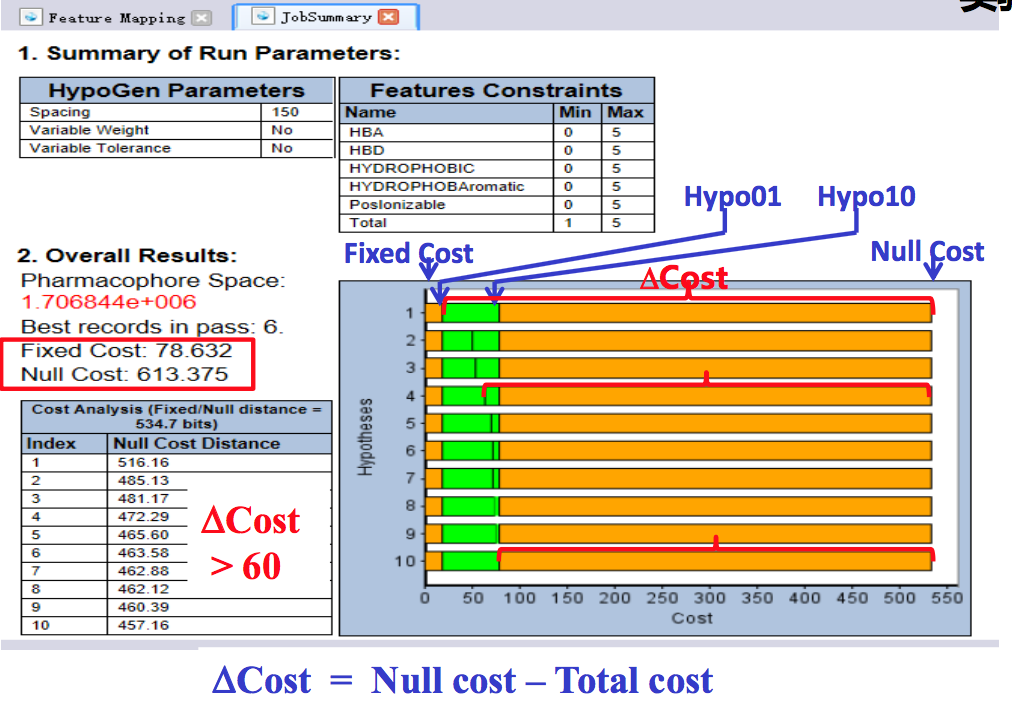
△Cost值判断模型置信度
Null cost :
- 假设一个模型没有任何药效特征元素,该模型预测的化合物活性值即平均活性值,该 模型的cost值即null cost值;
- 在药效团模型产生过程中该cost值最大,是模型cost值的上限。
Fixed cost:
- 理想模型的cost值,该模型最简且预测的化合物活性值同实验活性值无偏差或偏差极小,该模型的cost值即fixed cost值;
- 在药效团模型产生过程中该cost值最小,是模型cost值的下限。
Total cost:
- 产生的每个药效团模型的cost值,是configuration cost,error cost 和weight cost三者乘以一个系数(默认为1)的加和。
△Cost = Null cost – Total cost

4.3 其他资料
模型构建
-
选择一组训练配体。 选择一组结构多样的分子,用于开发药效团模型。由于药效团模型应该能够区分具有和不具有生物活性的分子,该组分子应该包括活性和非活性化合物。
-
构象分析(Conformational analysis) 。 产生一组低能构象,这些构象可能包含每个选定分子的生物活性构象。
-
分子叠加(Molecular superimposition)。 叠加(“拟合”)分子的低能构象的所有组合。可以拟合该组中所有分子共有的类似(生物等排的)官能团(例如,苯环或羧酸基团)。推导出最佳拟合的一组构象(来自每个活性分子的一种构象)被认为是活性构象。
-
抽象(Abstraction )。 将叠加的分子转换为抽象表示。例如,叠加的苯环可以更概念地称为“芳环”药效团元素。同样,羟基可以指定为“氢键供体/受体”药效团元素。
-
验证(Validation)。 药效团模型是一种假设,它解释了一组与常见生物靶标结合的分子的生物活性。该模型仅在能够解释一系列分子的生物活性差异的范围内才有效。
随着新分子的生物活性变得可用,可以更新药效团模型以进一步改进它。
五、具体案例
5.1 训练集分子的准备
[Files Explorer]-[Samples | Tutorials | Pharmacophore]-双击 serm_ligands.sd • 在表格浏览器中,点击 (向上)和 (向下) 键,观察各个分子的3D结构特征。
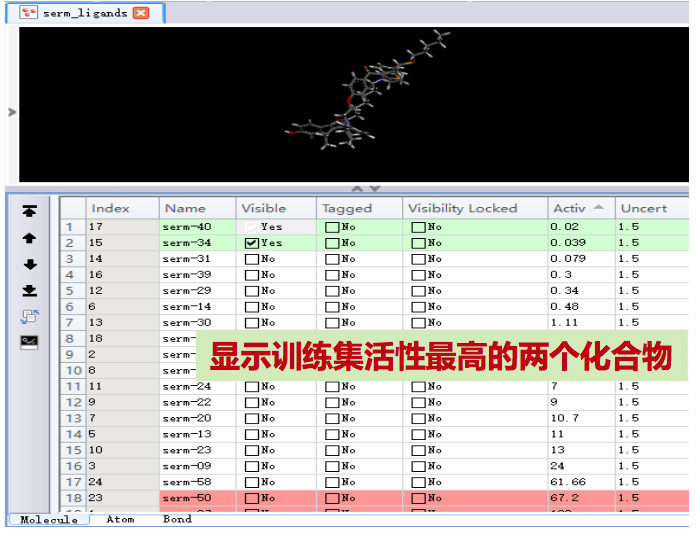
5.2 药效特征元素的提取
- 在表格浏览器中,右击鼠标并选择Color By Activity。
- 双击Activ一栏,使24个化合物 按活性从高到低(IC50 值从低到 高)排列。
- 点击 (向上),显示活性最高的化合物 (serm-40);勾选活性排名第 二化合物(serm-34)的Visible 复选框。
活性化物:MA * Unc ^(MA) - A /Unc^(A) > 0.0
非活性化物: log(A)-log(MA)>3.5

识别所选特征元素在活性最高的两个化合物中 所有可能的位置

[Edit and Cluster Features]-[Current Features:All Features]:查看训练集活性最 高两个分子所表征的药效特征元素的所有类型

5.3 3D QSAR药效团的构建
测试集分子的准备: [Files Explorer] - [Samples | Tutorials | Pharmacophore],双击打开 test_serm_ligands.sd
[Tools Explorer]-[Pharmacophores | Create Pharmacophores Automatically]-[3D QSAR Pharmacophore Generation…]
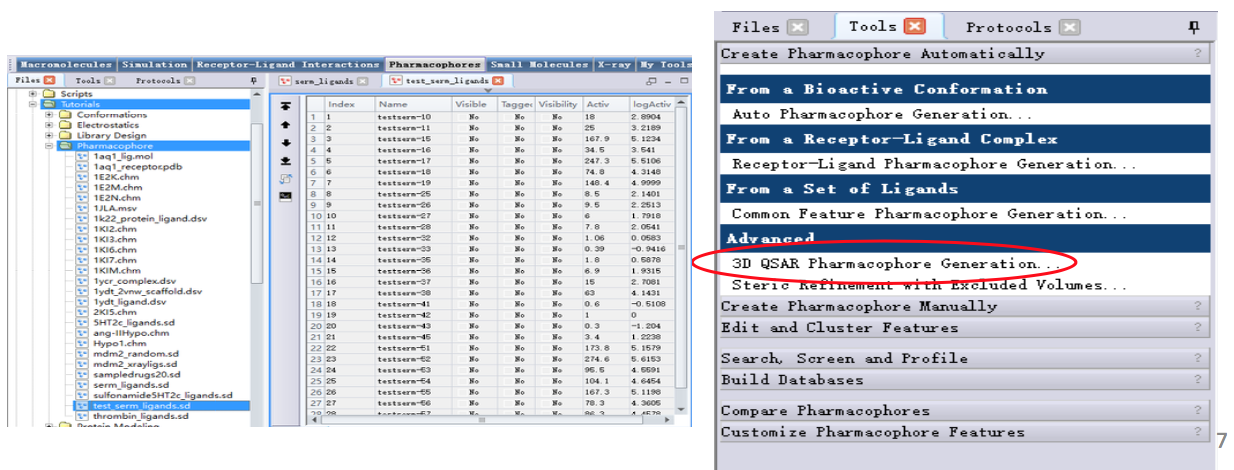
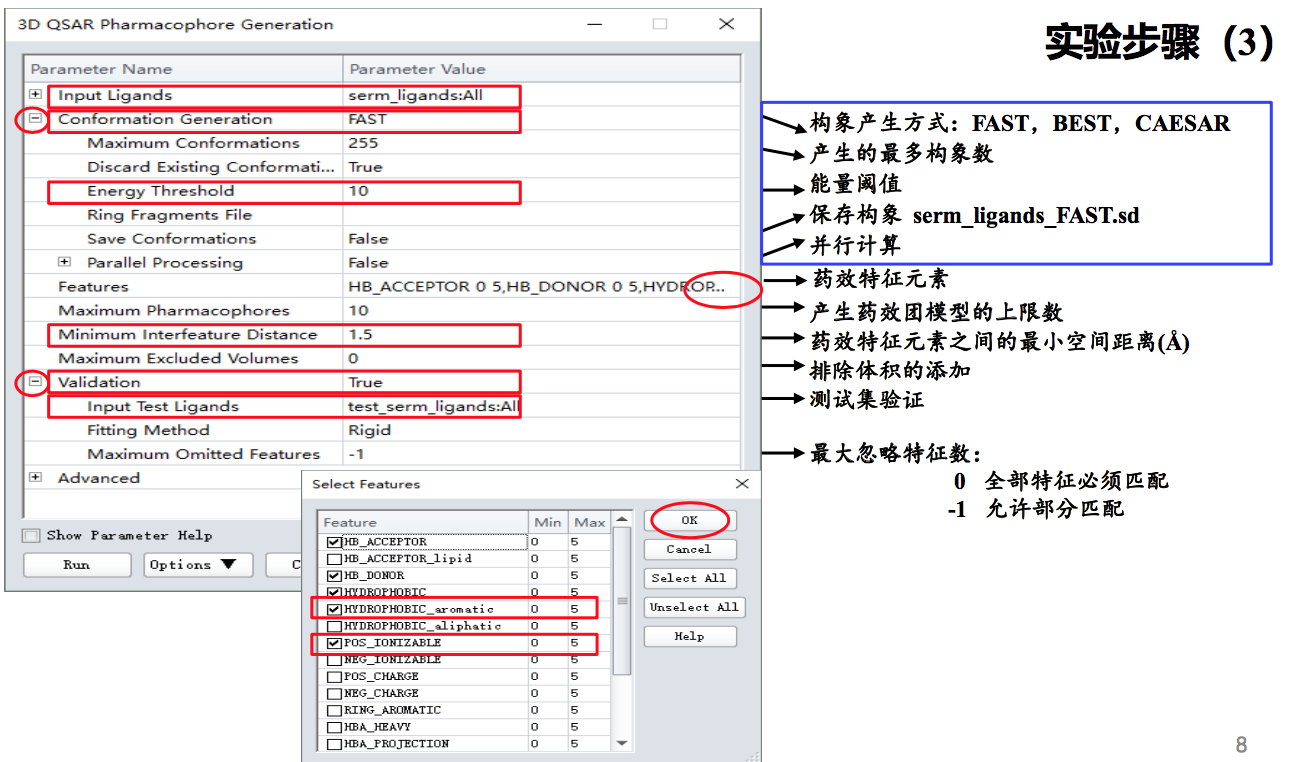
其他参数设为默认
点击Run运行作业
点击Background等待作业完成
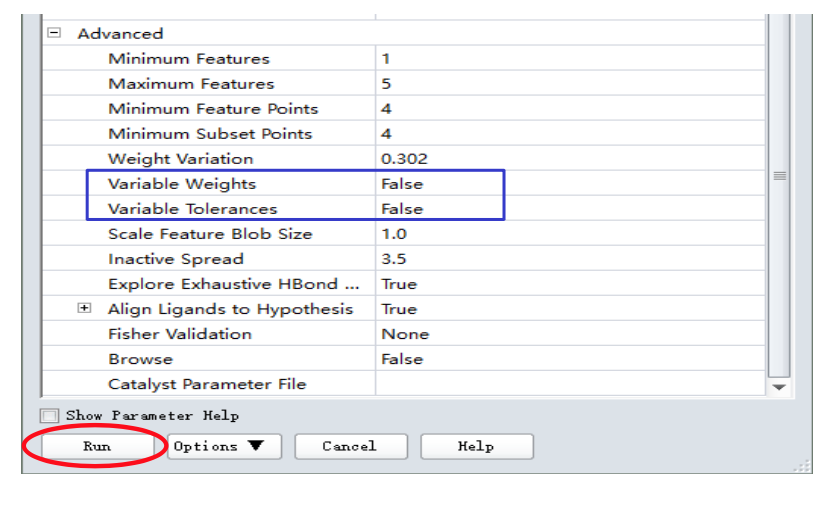
5.4 药效团结果分析
结果查看:[Jobs Explorer]-双击[3D QSAR Pharmacophore Generation]对应的行。
收缩Summary目录
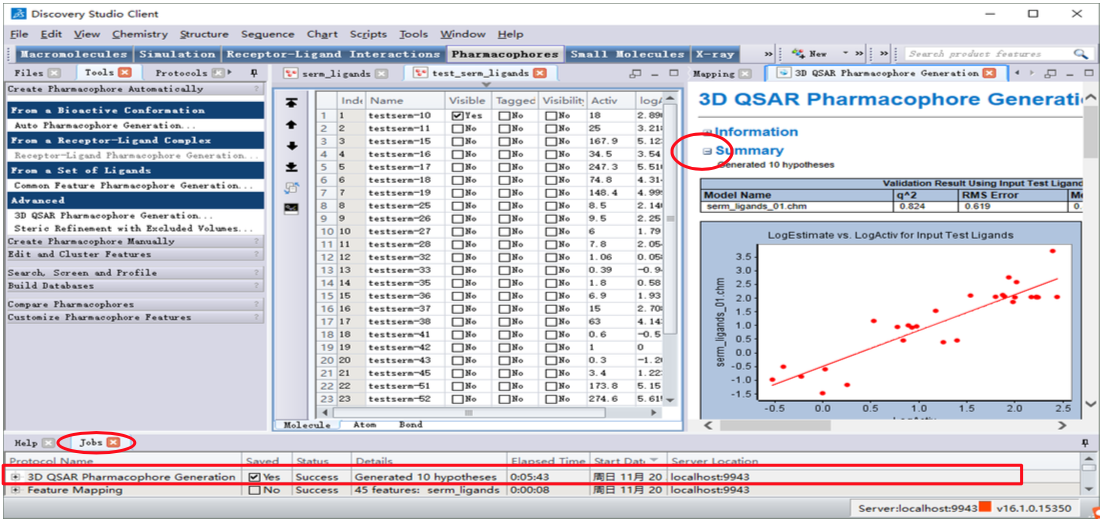
(1)训练集分子与模型的匹配
- 展开[Results]
- 点击链接:[View Aligned Ligands 01] , 查看训练集分子与排 名第一的药效团 Hypo1(serm_ligands_0 1)的匹配情况及活性值 预测情况

点击(向上)显示预测活性最高的化合物 (serm-40) 与药效团 serm_ligands_01的匹配情况
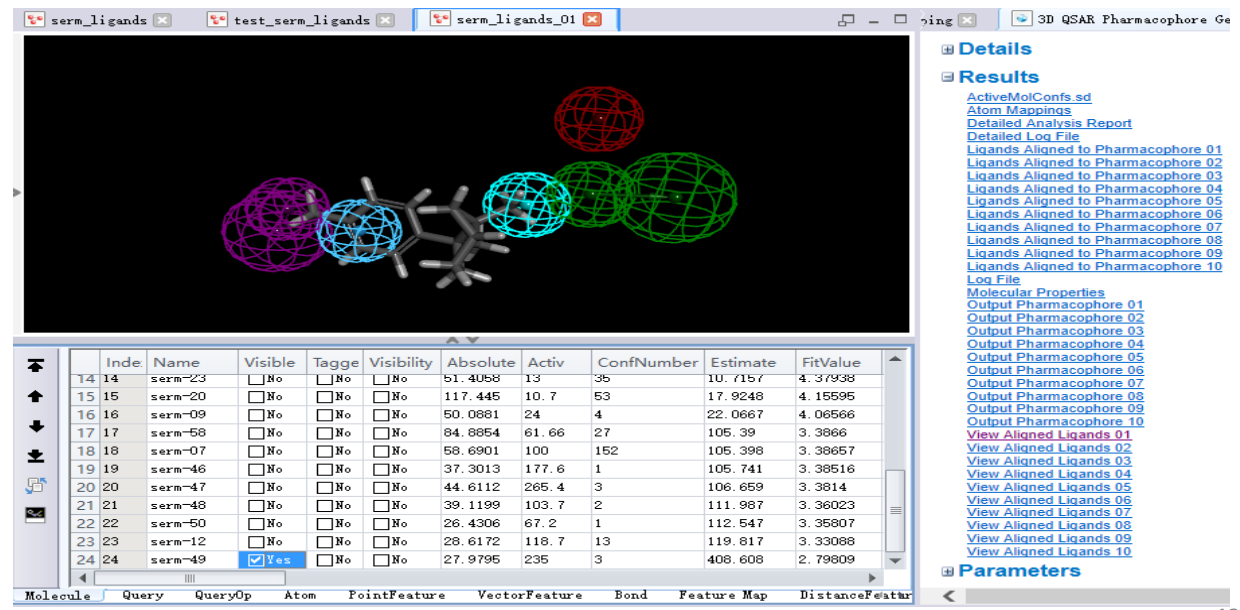
点击 (向下) 显示预测活性最低的化合物 (serm-49) 与药效团 serm_ligands_01的匹配情况

(2)Log文件分析
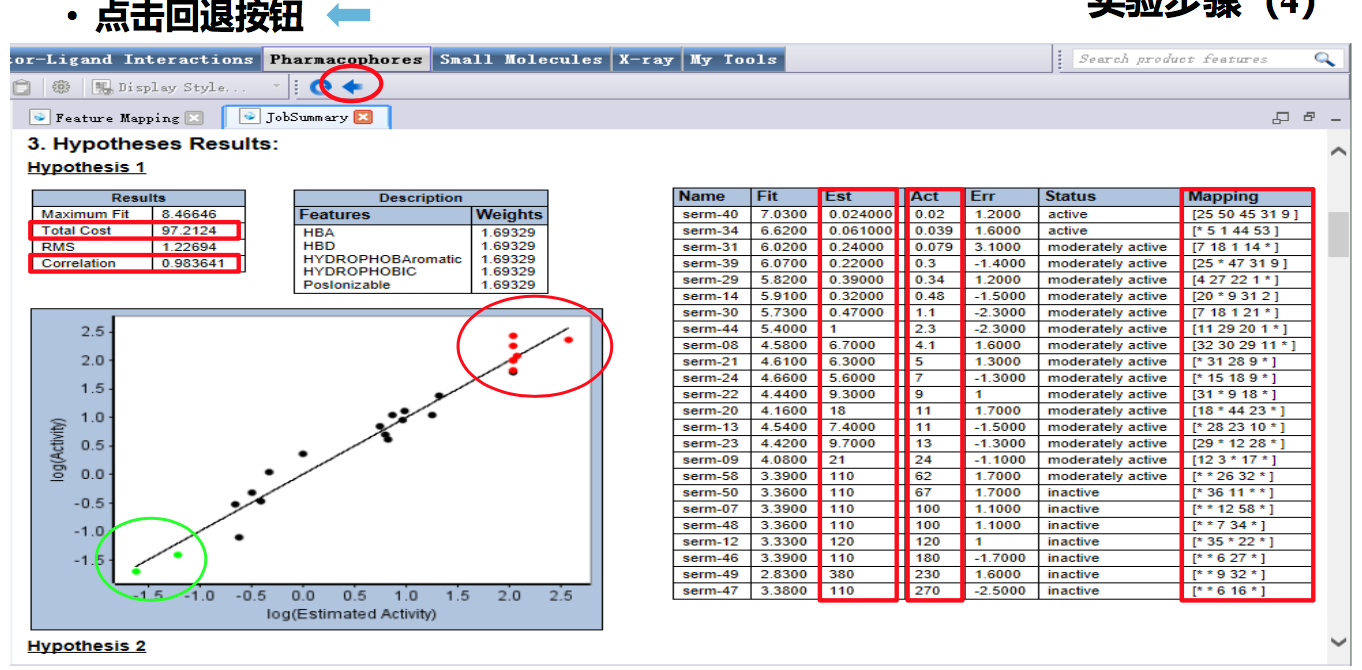
点击[Result]-[Detailed Analysis Report]打开cost分析报告。
最大化右侧窗口
- 第一部分:参数设置
- 第二部分:整体模型评估
- 第三部分:单个模型评估

(3)测试集验证
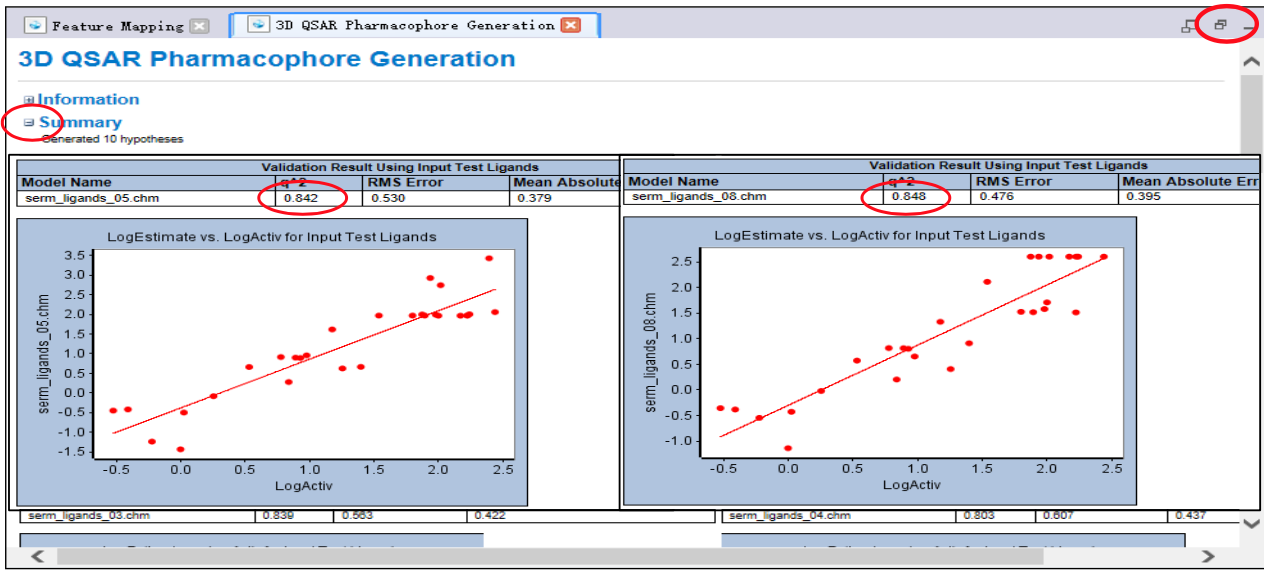
- 展开[Summary]
- 参看完毕后,还原右侧窗口大小
5.4 活性预测
- [File Explorer]-双击打开桌面文件 predict_serms_ligands.sd
- [Tools Explorer]-[Pharmacophores | Search, Screen and Profile]-[Ligand Pharmacophore Mapping…]
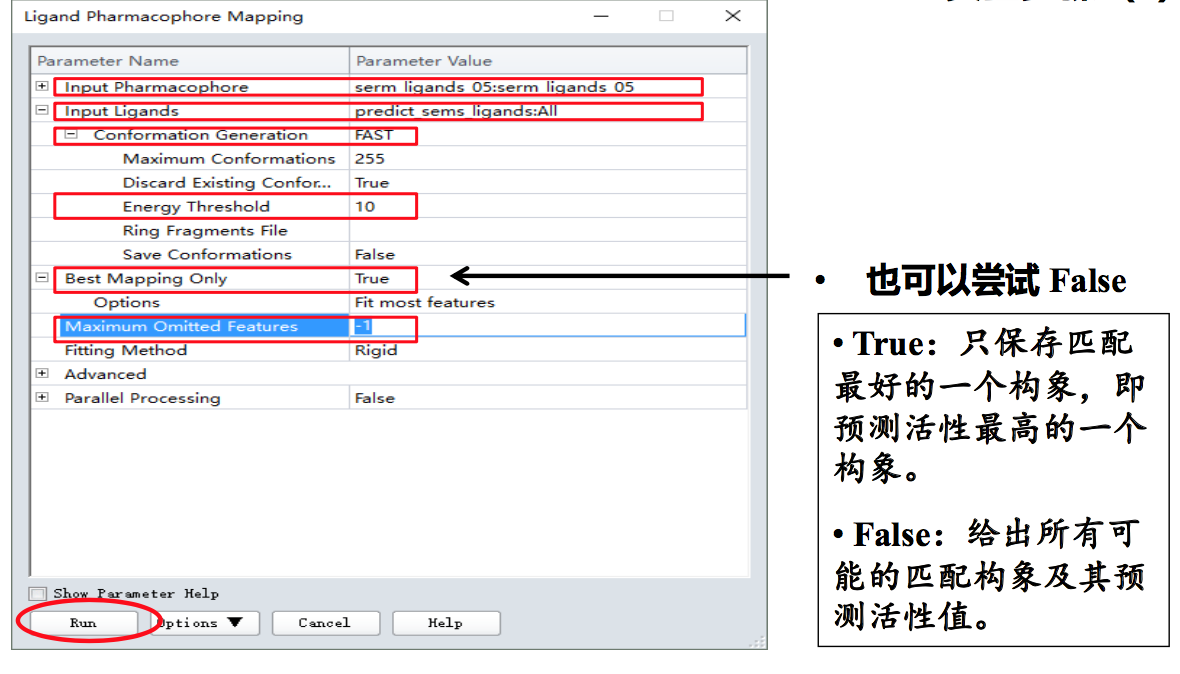
- [Jobs Explorer]-双击[Ligand Pharmacophore Mapping]对应的行;
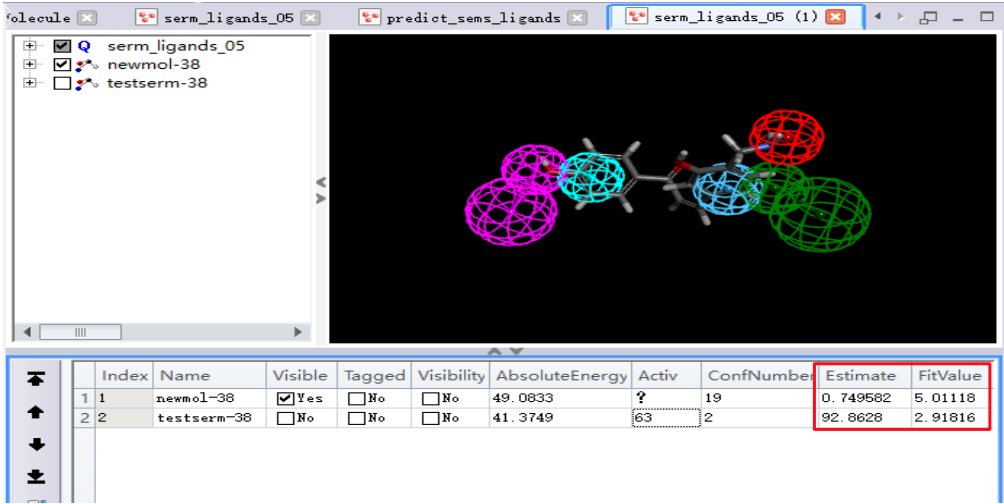
- 点击[View Results],查看预测活性值Estimate和FitValue值。
参考资料
- https://en.wikipedia.org/wiki/Pharmacophore
- 《基于药效团的药物设计方法和应用》 https://max.book118.com/html/2017/1203/142584736.shtm
- 《基于药效团的药物发现》 https://max.book118.com/html/2015/0819/23721333.shtm
- 复旦大学 谢琼 老师的 《药物设计学》 课件
