【3.7.1】变构药物(Alloseric drug):从偶然发现到基于结构的合理设计
近年来,变构药物设计与发现(Alloseric drug design and discovery)在药学领域同仁的努力下,从被认为难靶只能偶然获得到开始合理的药物设计,取得了长足的进步。与靶标正构位点(Orthosteric site)的调节剂相比,调节剂结合在结构多样的变构位点(Allosteric site),比正构调节剂天然拥有更好的靶向性。然而,由于变构调节性质复杂,10年前,已发现的变构调节剂基本都是偶然获得的。随着近10年对于变构数据的系统积累和随之的变构调节机制理解深入,我们与国内外同道一起推进了变构药物设计方法的发展,进而有效指导了基于结构的变构调节剂发现过程。今天,和大家一起回顾一下10年来变构概念的演变,并讨论新药研发中变构调节剂的优点和其发现中的障碍。下次,我们将着重叙述当前在变构研究领域中可用的药物设计方法和实验方法,及两种方法配合使用的一些成功变构药物先导研究案例,为变构药物设计方法在更多孤儿受体靶标上获得原创先导化合物突破提供研发线索和可行性。
一、引言
变构(Allostery)是生物大分子的本质属性。对蛋白质来说,这种属性意味着一个区域(如变构位点,Allosteric site)可以与远距离的另一个区域的运动/能量相协同,并传递蛋白内信号(变构通路,Allosteric pathway)。通常,变构信号从变构位点到具有功能的正构位点的传播是由变构位点的扰动引起的,这种扰动包括调节剂(离子、小分子、蛋白质、DNA和RNA)结合,点突变和翻译后修饰。变构调节在生理状态下通过变构位点影响生物大分子的功能,进而实现对无数生物过程的精准调控,包括从酶催化,基因表达,细胞分化到新陈代谢与机体平衡。由于对机体生命过程的变构控制无处不在,变构调节被认为是生命的第二秘密,成为洞察细胞生理和病理命运的重要方式。
从创新药物研发的角度来看,蛋白变构为靶标新型药物的出现提供了极具吸引力的前景。由于变构位点和正构位点(Orthosteric site)不处在同一位置,变构调节剂结合变构位点可上调和下调蛋白功能,并不和保守正构位点上的内源配体竞争,这一特点提供了激动型药物的普适开发方式。而且,相对于高度保守的正构位点,多样化的变构位点赋予了变构激动剂/抑制剂更好的选择性和更低的毒性。更为重要的是,蛋白中的变构抑制剂和正构抑制剂联用还可以对蛋白功能进行协同抑制,来降低药物耐药突变产生的可能。
尽管利用变构药物具有如此巨大的优势,但变构先导化合物的发现一直是原创药物发现领域的重大瓶颈。2010年以前,大多数报道的变构分子都是通过高通量筛选实验偶然发现的,药物开发者缺乏对变构药物先导化合物及其对蛋白靶标分子机制的影响给予预期和设计。随着Allosteric database( ASD,http://mdl.shsmu.edu.cn )的建立,有关变构蛋白、变构位点及其调节剂的数据可用性与日俱增。为建立合理识别变构位点并筛选变构机制药物发现方法的发展提供了可能。近10年来,随着相关方法的建立和不断完善,面向新靶标从头结合药物设计方法和实验方法发现变构调节剂的开发已经出现。
二、变构概念的过去和现在
Bohr在1904年的开创性工作揭示了一种变构现象,即血红蛋白和二氧化碳的结合影响氧与血红蛋白的结合亲和力。这种人尽皆知的现象描述了分子与两个不同位点的协同结合,它曾经被称为“玻尔效应”,目前被称为“变构效应”。1961年,Monod和Jacob首次提出了变构的概念,以解释酶活性的反馈抑制机制,其中关键在于“抑制剂不是底物的空间结构类似物”。后来,在20世纪60年代,研究者们创建了两个经典的变构相互作用模型来解释变构分子的协同结合:一个被称为“协调模型”或MWC(Monod-Wyman-Changeux)(1965)模型。另一个是’序列模型’或KNF(Koshland-Nemethy-Filmer)(1966)模型。两种模型都强调了构象变化在变构调节终态结构中的作用。
1984年,Cooper和Dryden提出了一个名为“动力学驱动的变构”的理论模型,该理论模型认为,变构效应发生时,平均结构中不会出现明显的构象变化,这揭示了熵对变构的重要性。核磁共振(NMR)光谱学技术的发展更好地探究了蛋白质的内部运动,进而对探测“动力学/熵驱动的变构”有很大帮助,例如在分解代谢物激活蛋白(CAP)、PDZ结构域、钙调蛋白和蛋白激酶A中。最近,Frederick等人发现了蛋白质构象熵与总结合熵的变化呈线性关系,揭示了构象熵在蛋白质识别中的重要作用。他们进一步发展了一种通过侧链的运动和蛋白质构象熵的变化来经验地,定量地描述构象动力学变化之间的关系。
1999年,Nussinov及其同事提出了一种“构象选择和转移”模型,该模型从自由能变化的角度解释了变构的运作方式。这种模型将变构的概念从两种结构状态扩展到多种结构状态的构象集合。在同一年,Ranganathan及其同事提出了“变构网络”模型,该模型假设通过物理互连残基的网络实现蛋白质不同位点之间的信号传递。这种观点已经被大量高分辨率显微结构证实。最近,Hilser及其同事提出了一种“集合变构”模型来解释变构的起源。Hilser的模型在解释变构起源时类似于Nussinov的模型。两种模型都假设蛋白质集合的所有可能构象都拥有其各自的能量,而变构分子与蛋白质的结合重塑了蛋白自由能分布。然而,Hilser的模型进一步扩展了Nussinov的模型,以解释本质上无序蛋白质中的变构;它提供了一个统一了结构化的、动态的和无序的系统中的变构效应的框架。
这段历史显示,变构概念的提出和模型的建立已经超过50年,因为“变构”这个词是由Monod和Jacob创造的。很明显,变构的概念已经从两态模型和静态结构模型演变为动态集合模型。当前这个现象被用于药物开发,重要的是,基于这种现象的药物设计方法已经可以通过合理的识别变构位点和突变,发现变构激动剂和抑制剂,评估变构相互作用,并研究变构机制,所有这些都将在下次详细阐述。
三、变构调节剂发现的优势和挑战
3.1.优势
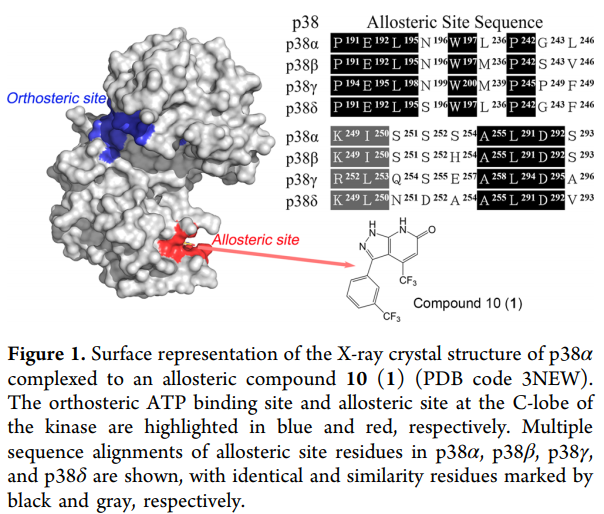
与占据蛋白质活性位点的正构调节剂相比,变构调节剂具有几个明显的优势。这些优势可以通过它们的高特异性和低副作用来证明。G蛋白偶联受体(GPCR)和蛋白激酶是治疗药理学中两个最重要的药物靶标。高度进化保守的内源性正构结合位点是开发用于GPCR和蛋白激酶的选择性正构药物的关键挑战。已经认识到,GPCR或蛋白激酶的预期正构调节剂经常与其同源蛋白质发生交叉反应,导致意料之外的副作用和脱靶毒性。拥有着多样的结构和拓扑学上的位置差异,作为替代方案的变构位点使人们能够克服正构调节剂遇到的这两个主要障碍。例如,基于吡唑并吡啶的化合物10(1)与激酶C-叶的变构位点结合。由于该位点内的残基差异,对四个密切相关的结构而言,该化合物对p38α具有极好的选择性(IC50:1.2μM),对p38β,p38γ和p38δ的活性值都不高(IC50:>40μM)(图1)。
在时间和空间方面上,变构调节剂具备特异性的一定基础。它们与内源性配体一起通过微调蛋白质功能,而不是关闭或打开内源性生理信号通路来发挥它们的协同作用。这一特征可以在提高了过量使用变构药物时的安全性。
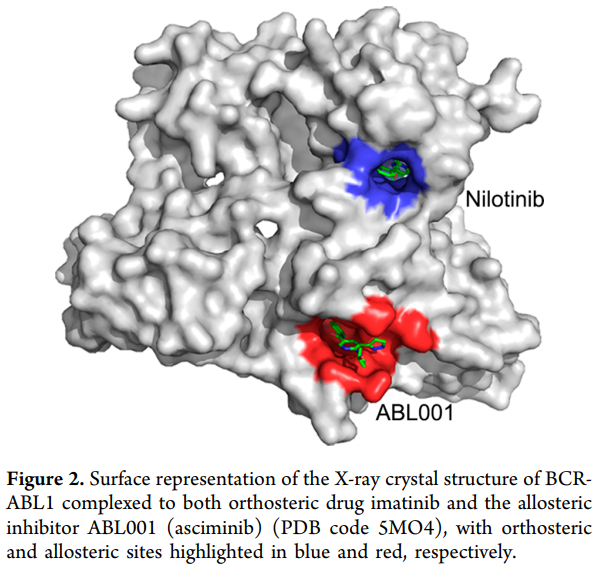
重要的是,变构调节剂具有对抗位于患者的正位位点的耐药突变的潜力。例如,BCR-ABL1的’看门人突变’T315I引起对一组临床批准的正构药物的耐药性,如伊马替尼,博舒替尼,尼洛替尼和达沙替尼。利用变构抑制剂ABL001(asciminib)对T315I突变激酶C-叶的肉豆蔻酰口袋的治疗可克服其耐药性(图2)。
蛋白质存在于由多种不同的构象状态组成的整体中,这些构象状态可能参与多种信号传导途径。与蛋白质独特构象结合的变构调节剂可以实现具有偏向性的信号传导。例如,G蛋白或β-arrestin介导的GPCR下游信号级联就可以通过受体-配体复合物的不同构象状态激活。
值得注意的是,变构调节还为药物治疗难以靶向的“无可救药的”靶标提供了新的治疗希望。这类蛋白质由于底物结合强,缺乏深的结合位点或蛋白质-蛋白质相互作用(PPI)界面大而扁平而对难以药物产生相互作用。在这些情况下,变构调节剂可以绕过与正构位点或PPI界面的直接竞争,并结合替代的位点来微调蛋白质活性。此外,如果明显的结合位点在解析的晶体结构中难以寻觅,那么可以从蛋白质的次要或中间构象中捕获隐蔽的(或隐藏的)变构位点,进而用于药物设计。目前,许多以前被认为是“无法靶向”靶点已被证明可通过变构调节方法治疗,包括K-Ras,转录因子(MYC和NF-κB,BCR-ABL的SH2激酶结构域PPI界面,磷酸酶(SHP2, PTP1B和PP2A),缺氧诱导因子-2和STAT3。
近年来变构调节剂数量的爆炸性增长已经证明了在药物发现中探索变构效应的优势。然而,变构调节剂的发现也存在着重大挑战,可以通过更深入理解变构机制的分子细节,变构蛋白和调节剂的结构和生化特性来迎接这一挑战。

3.2.挑战
变构调节剂发现的关键步骤是鉴定蛋白质中的真正的变构位点。与占据保守的已知正构位点的正构调节剂不同,变构位点上的进化保守性较低。理论上,蛋白质在空间上不同于正构位点的任何结合口袋都可视为潜在的变构位点。此外,难以确定潜在变构位点对正构位点的调节作用。这些缺点长期且严重地阻碍了变构调节剂的发现过程。幸运的是,近年来由ASD和Asbench整理的X射线晶体学和核磁共振光谱学提供的大量变构蛋白质-变构分子复合物部分地解决了这一困境。基于ASD和Asbench已经开发了许多药物设计方法来识别变构位点,极大地促进了基于结构的变构调节剂的发现。
挑战也可能源于在变构部位出现的耐药性突变,这与正构药物所面临的情况相同。例如,在表达BCR-ABL1变体的Ba / F3细胞系中,在激酶的C-叶中的变构肉豆蔻酰基位点处的单点突变(A337V,P465S,V468F和I502L)赋予该蛋白对变构抑制剂ABL001的抗性(图3)。此外,ABL001和正构药物尼洛替尼的组合比单独使用每种药物更有效地抑制突变体。这种加和效应表明变构和正构药物在人类疾病治疗中进行协同作用是可能的。大量证据表明,与正构位点相比,变构位点在较低的进化压力下进化,这意味着变构突变的发生频率高于正位突变。实际上,由于进化上较不保守的变构位点,已观察到丙酮酸激酶在变构调节剂结合域中有20多种与疾病相关的突变。
此外,突变可发生在变构位点以外的变构通讯途径,改变蛋白质动力学并对变构调节剂产生间接抗性。因为蛋白质中的变构网络的复杂性,这种突变很难通过实验鉴定。此外,由于变构位点的低进化保守性,变构调节剂的效应显示出明显的物种差异。例如,变构候选药物的作用在重组的人类体外受体中是有效的。然而,当使用啮齿动物受体或模型来检查药物效应时,可能获得相反的结果。一般而言,变构调节剂具有比正构调节剂更低的结合亲和力,并且经常遇到“平坦的”难以发生相互作用的结构-活性关系。此外,变构调节剂通常具有比正构配调节剂更低的水溶性。这些性质不仅导致变构调节剂的临床实验难以实现,也影响了变构蛋白质-调节剂复合物的结晶 。
四、变构调节剂发现的现状
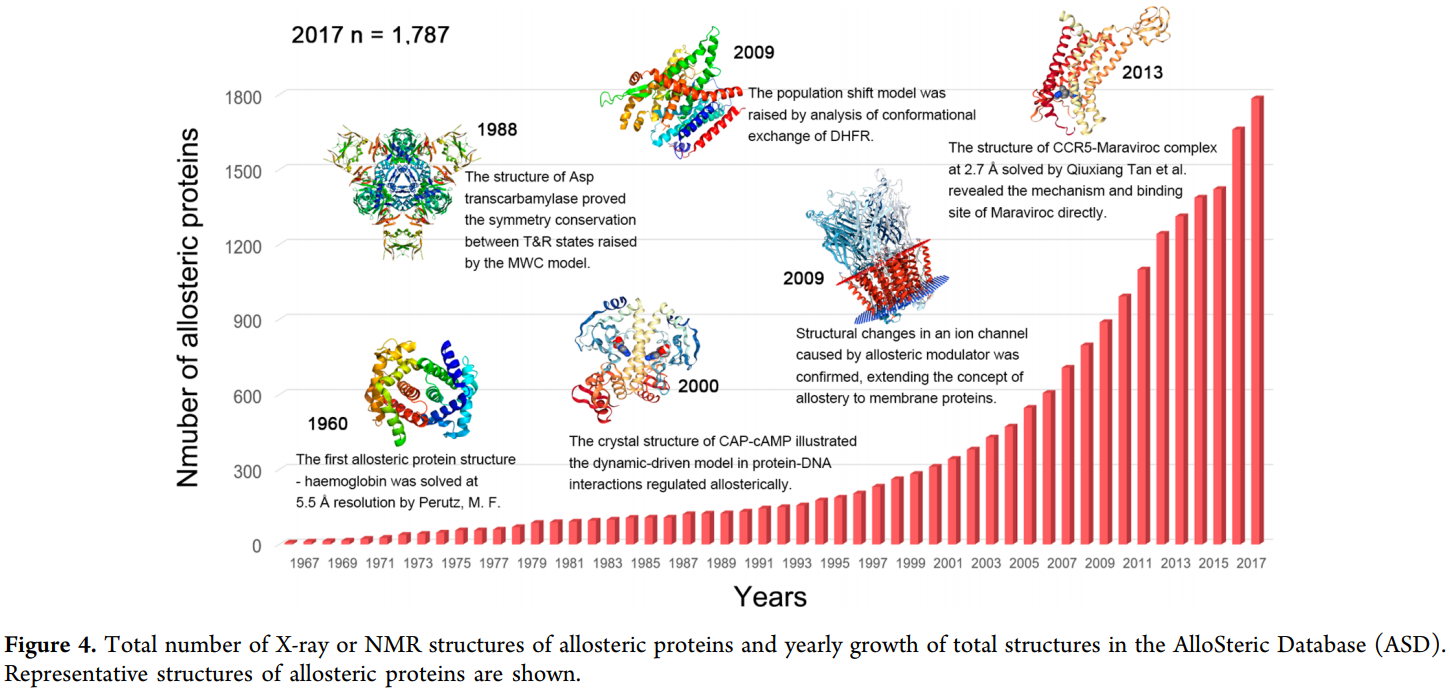
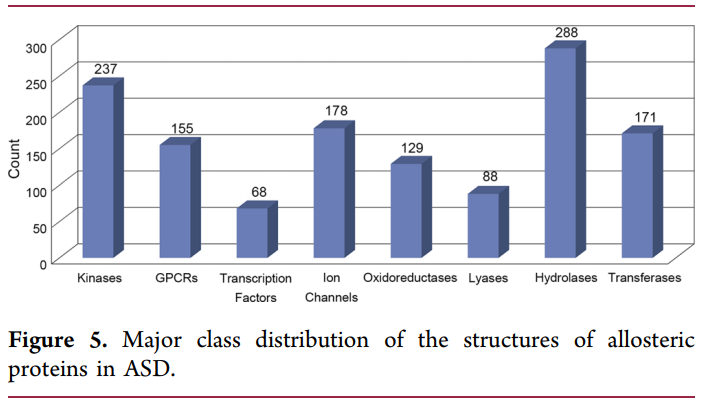
截止到2018年12月,根据变构数据ASD( http://mdl.shsmu.edu.cn )统计,当前约1,800个各种来源的变构蛋白(图4),它们主要分布在8个蛋白质家族中:激酶,GPCR,转录因子,离子通道,氧化还原酶,裂解酶,水解酶和转移酶(图4)。在变构研究的最初阶段,只在以血红蛋白为原型的多聚体蛋白中观察到变构调节现象。由于化学和结构生物学技术的进步,目前已经在如GPCR,激酶和酶的单体蛋白质中观察到变构效应,这极大地扩展了可用的变构药物靶标集合。
近年来,由于国际各大药物发机构对变构调节剂研究的不断增加,变构调节剂的数量迅速增加(图5)。ASD现在包含80,000个变构调节剂,靶向蛋白质超过1300种。五种变构调节剂已被美国FDA批准为市售药物,包括三种GPCR(Cinacalcet®,Maraviroc®和Plerixafor®)和两种激酶(Gleevec®和Mekinist®)变构药物。此外,许多有希望的候选药物正在进入临床试验。
图6.ASD中变构调节剂的年增长率。市售的变构药物的化学结构如图所示。
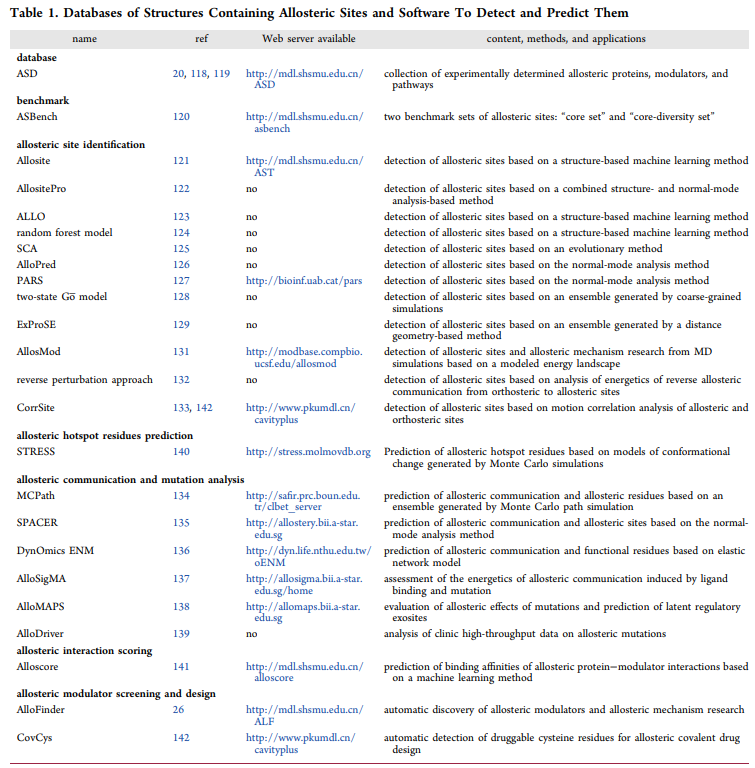
由于变构相互作用的复杂性质,使用诸如X射线晶体学和NMR的实验方法确定蛋白质中的变构位点和该变构的机制通常会耗费大量时间,且结果不总是令人满意。作为实验方法的可行替代方法,发展研究变构的药物设计方法成为变构药物发现突破的重要手段。我们总结了代表性的变构药物设计方法及其当前可用性如下:

总的来说,随着变构药物设计方法的发展和不断突破,基于已知蛋白质靶标进行变构调节剂设计方面取得了一些重要进展。这些技术与理解变构激活或抑制的实验方法的进步相结合,将成为未来新靶标药物发现的重要利器和常规手段。
参考资料
- Lu S, HeX, Ni D, Zhang J*. Allosteric modulatordiscovery: from serendipity to structure-based design. J Med Chem. 2019, doi: 10.1021/acs.jmedchem.8b01749.
