【5.8.1】PROTACs简介
靶向蛋白水解的嵌合体(PROTAC,Proteolysis-targeting chimeras)和相关分子通过泛素-蛋白酶体(ubiquitin–proteasome )系统诱导靶向蛋白降解,代表了一种新的治疗方式,并且是人们关注的焦点,因为与传统的基于位置的抑制剂相比,在剂量,侧面方面具有潜在优势 效果,耐药性和调节“非药物”目标。 但是,该技术仍在日趋成熟,目前正在阐明成功使用基于PROTAC的药物的设计要素。 重要的是,到目前为止,在600多种E3泛素连接酶中,只有不到10种被用于靶向蛋白质降解,而扩大这一领域的知识是一个关键的机会。 在这里,我们简要讨论了在化学生物学和药物发现中有关靶向蛋白质降解的经验教训,并系统地审查了人类E3连接酶的表达谱,结构域结构和化学易处理性,这可能会扩展PROTAC发现的工具箱。
一、前言
尽管有效和选择性的小分子蛋白功能抑制剂的开发不断取得进展,但是对于典型的小分子药物而言,具有高生物医学相关性的多个靶标仍然具有很高的挑战性。 另外,尽管诸如单克隆抗体和寡核苷酸疗法之类的生物学方法可以提供解决此类靶标的机会,但它们具有局限性,例如递送受限。 因此,最近的进展表明,用小分子药物进行靶向蛋白质降解可能成为一种新的治疗方式,引起了人们的极大兴趣。
蛋白质降解是细胞内蛋白质更新的正常过程。它提供了蛋白质折叠过程中的质量控制机制,快速响应变化的细胞信号的能力以及调节可用氨基酸库的机制。大多数蛋白质将通过泛素蛋白酶体系统(UPS,ubiquitin–proteasome system)降解。尽管该过程足够广泛以涵盖蛋白质组的广泛多样性,但它仍然通过一系列受调控的步骤来运作,在这些步骤中,蛋白质被泛素进行共价翻译后修饰而被标记为降解。蛋白质的泛素化是通过三种酶的级联反应进行的。
- 第一步,ATP被E1泛素激活酶消耗,产生活化的泛素-腺苷酸,该蛋白通过与E1活性位点中的催化半胱氨酸共价连接而转化为硫酯中间体。
- 随后进行转硫醇化反应,其中泛素从E1酶的催化半胱氨酸转移至E2(泛素结合)酶的催化半胱氨酸。
- 最后,遍在蛋白通过桥接E3遍在蛋白连接酶的作用转移到底物蛋白上,在泛素的羧基末端和靶标的赖氨酸侧链之间形成异肽键。
可以重复该循环以产生多聚泛素链,该泛素链指导底物在蛋白酶体处降解。在这种级联反应中,E3连接酶在决定靶标特异性方面的作用是独特的。 E3连接酶代表着约600个预测成员的大基因家族,通常起衔接子分子的作用,可通过蛋白质-蛋白质相互作用识别底物,并通过将那些靶标保持在相关的泛素化机制附近来促进泛素化。
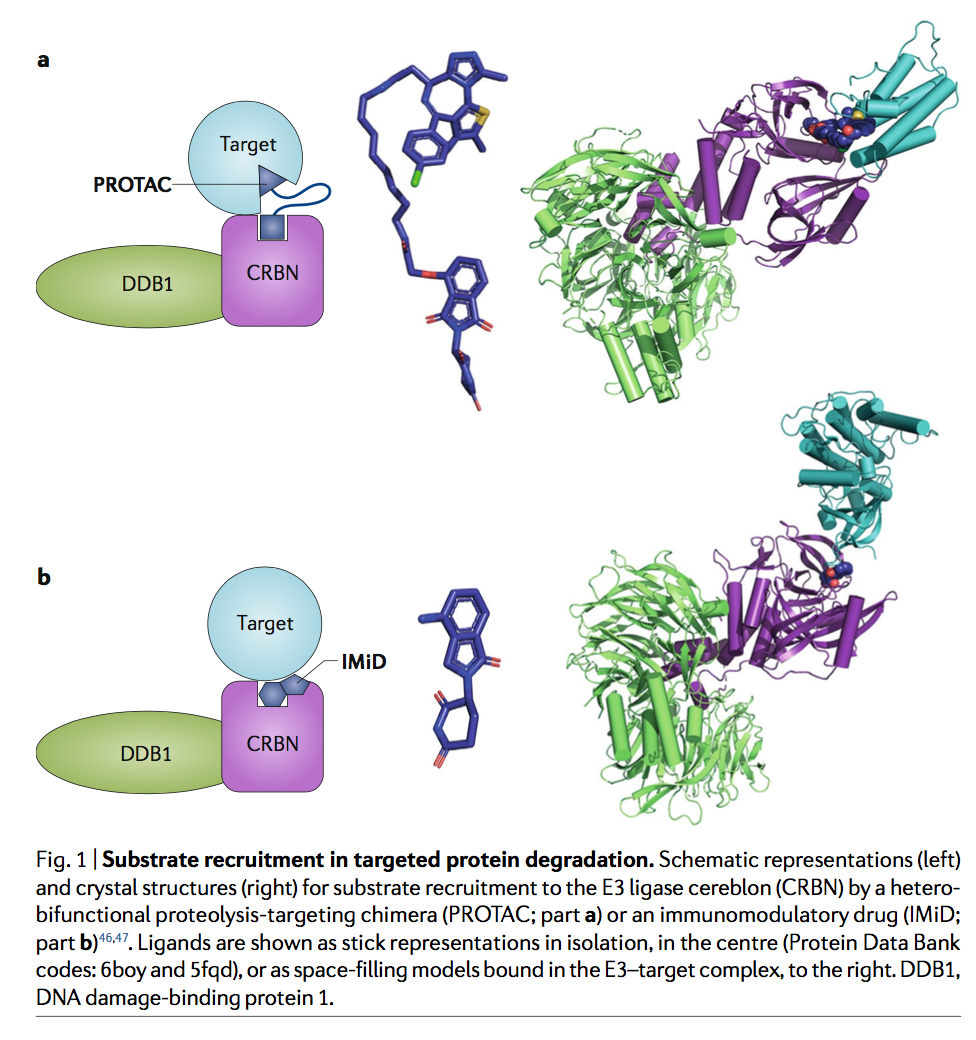
通过E3连接酶BTRC,在近20年前就证明了通过人为招募E3连接酶来重定向蛋白质降解的潜力。在这项工作中,产生了一个包含与BTRC的肽配体连接的蛋氨酸氨基肽酶2(MetAP2)的共价配体的嵌合分子(衍生自IκBα的磷酸肽),该分子充当了靶标和E3连接酶之间的非天然“桥”(图1)。令人兴奋的是,这种依赖化合物的募集足以促进非洲爪蟾提取物中MetAP2的降解。尽管这项研究建立了令人信服的原理证明,但对这些“针对嵌合体的蛋白水解”(PROTAC)的兴趣仍然有限,直到此类分子从肽配体发展到完全合成的化合物为止。这首先在2008年证明了雄激素受体募集到E3连接酶MDM2(reF.3),此后不久又扩展到E3连接酶cIAP14。与此同时,Hiroshi Handa等人的独立研究表明,免疫调节药物(IMiD)沙利度胺直接与E3连接酶大脑(CRBN)结合,后来证明了这种结合事件可介导一系列依赖配体的降解通过一组IMiD类似物(包括来那度胺和pomalidomide)靶向目标(包括IKZF1,IKZF3和CK1α)。与上述PROTAC分子相似,IMid能够通过非天然E3连接酶募集来促进非天然靶标降解(图1)。
在过去的5年中,靶向蛋白质降解的领域已大大扩展,数十种示例性底物都适用于这种机制。 研究已经从细胞试验发展到对模型生物的体内研究,并且第一阶段临床试验的结果即将出现。 但是,仍然存在很大的挑战。 虽然募集E3连接酶是靶向蛋白质降解的必要步骤,但它似乎不足,因为PROTAC设计所有部分的微妙特征均可对降解潜能产生巨大影响。 另外,使用基于混杂激酶结合剂的PROTAC进行的蛋白质组学研究的新数据表明,某些靶标比其他靶标对这种作用机制的反应更敏感。 因此,目前蛋白质降解领域需要大量的经验筛选。 确实,即使在可降解的靶标中,E3配对的选择也显得至关重要。
我们建议该领域的关键推动力是对整个E3连接酶基因家族的更广泛探索。 确实,迄今为止,在靶向蛋白质降解方面仅研究了600个家族成员中的约1%。 在本文中,我们讨论了靶向蛋白质降解领域的机遇与挑战,并综述了可用于扩展PROTAC工具箱的E3连接酶的主要类别。 我们讨论了E3连接酶配体性的特征,但请注意,即使具有中等E3连接酶亲和力的PROTAC也已证明能够指导有效降解。 如果完全实现,这种方法具有巨大的潜力,可以以新的方式追求既定目标,并通过靶向目前不适于抑制的临床相关蛋白来扩展可药用基因组。
二、进度和经验教训 Progress and lessons learned
最近的观察表明,IMiD和PROTAC有望在未来几年内对药物发现产生重大影响(表1)。 IMiDs的临床功效至少部分受其诱导CRBN介导的新底物降解的能力驱动:癌细胞对多发性骨髓瘤IMiD药物lenalidomide具有耐药性,因为IKFZ3中的单个氨基酸发生突变从而挽救了该转录因子来自蛋白水解降解; CK1α的单倍表达不足使骨髓增生异常综合征(MDS)细胞的染色体5q(del(5q))缺失对lenalidomide对MDS(5q)患者的CRBN介导的CK1α降解的敏感性;在2a期研究中,CRBN调节剂C-220的治疗比先前的IMiDs更能诱导IKFZ1和IKFZ3降解,在全身性红斑狼疮患者中引起阳性反应。临床前和临床研究证明了多种给药途径,包括口服。已经证明了PROTAC在器官和组织中的广泛分布,包括最近在大脑中的分布(Arvinas新闻稿;请参阅相关链接),而针对肿瘤学目标的PROTAC可以在异种移植模型中诱导持久性应答。对激酶抑制剂依鲁替尼( ibrutinib )有抗药性的癌细胞,对依鲁替尼衍生的PROTAC的治疗有反应,表明PROTAC可用于解决影响亲本抑制剂的耐药机制。最后,最近第一个将雄激素受体募集到E3连接酶的异双功能PROTAC进入了临床。

除临床或临床前数据外,化学生物学研究还强调了PROTAC作用机理的内在特性,可将它们与常规抑制剂积极区分开(表1)。 PROTACs由于其催化作用而不是基于占用的作用机制,因此可以实现较高的细胞效力,并且作用时间可以超出清除范围,并且取决于蛋白质靶标的周转率而不是停留时间。这些分子可以很好地靶向具有可配体结合口袋但不参与基因致病功能的蛋白质结构域[26,27],并且发现一些PROTAC比其来源的抑制剂更具选择性[12,28-30] ,可能是由于缺乏可接近的赖氨酸残基和/或去泛素酶的作用,并非所有接近给定连接酶的靶都被生产性泛素化。所选的E3连接酶的表达谱也可用于降解特定组织或细胞区室中的靶标。最后,对于由畸变形式的蛋白质(例如tauopathies)积累引起的疾病,目标降解而不是抑制是一种有前途的治疗方式。
还应注意PROTAC的开发带来的许多挑战。 对于临床应用,适当的剂量可能是个问题,因为饱和剂量的游离PROTAC分子可以拮抗二元PROTAC-蛋白质复合物与其三元伴侣的结合并消除催化降解, 一种有据可查的现象,称为细胞分析中的钩效应。 针对PROTAC治疗的获得性耐药性可以通过靶向E3连接酶复合物核心成分的基因组改变来驱动。 影响PROTAC结合但不影响蛋白质功能的突变也应该预期,当PROTAC利用功能中性位点时可能更是如此。 最后,迄今报道的PROTAC大于典型的口服小分子药物。 但是,现有数据表明,鉴于PROTAC的理化特性,其药代动力学的抑制作用可能不如预期的那样。
PROTAC作为化学生物学工具的发展也充满挑战。目前尚无指导特定E3连接酶与给定蛋白质靶标配对的理论依据:在PROTAC发现工作开始时尚不清楚,对接头化学的精挑细选的组合取样是否可以揭示接头长度和连接点的适当组合,这些组合对于生产性复合物的形成,或是否会浪费资源来尝试匹配无法配对的蛋白质。即使开发出具有出色选择性的PROTAC,决定其特异性的决定因素通常仍然复杂,晦涩或令人困惑。 PROTAC还可以降解其母体抑制剂无法触及的脱靶子,IMiDs可以降解各种ZnF转录因子阵列,并且已证明一些CRBN募集的PROTAC可以催化IMiD靶子IKFZ1和IKFZ3的降解。这些分子通常比典型的类药物化合物更大且更具柔韧性,这可以转化为膜渗透性差和对外排泵负责。虽然与E3连接酶的共价结合是可以接受的,但与蛋白质靶标共价结合的PROTAC可能会失去其机理的亚化学计量性质。最后,应降解目标蛋白以触发表型反应的百分比可能取决于靶标和读数(readout),但需要系统地进行研究。
三元复合物的最新晶体结构已经使人们对PROTAC的结构机理有了更深入的了解。对VHL和CRBN的结构研究表明,这些Cullin-RING E3连接酶(CRL)形成了大的,模块化的U形复合物,其中衔接蛋白介导了底物结合元件(VHL或CRBN)与结合了Cullin的Cullin支架之间的相互作用。 RING域蛋白RBX1,导致募集遍在蛋白结合的E2(图2a,b)。 U形导致E2和底物蛋白的近端定位,从而实现靶向泛素转移。这些大型复合物的结构有望提供一个扩展的泛素化半径,可以适应具有各种尺寸和形状的底物上的多个泛素化位点。特别是,考虑到衔接蛋白DNA损伤结合蛋白1(DDB1)的柔韧性,预计招募CRBN复合物的PROTAC降解的底物谱(可能还有其他DDB1-CUL4相关因子(DCAF)E3连接酶)由泛素化位点的可及性决定的较少,而由蛋白质合成速率以及连接酶-PROTAC-靶标结合事件的亲和力和动力学决定的更多。三元复合物的形成是由蛋白质-蛋白质和蛋白质-PROTAC相互作用驱动的,有时还包括稳定PROTAC接头与募集的蛋白质之间的接触(图2a)。

CRBN,目标蛋白含溴结构域蛋白4(BRD4)的第一个溴结构域与各种PROTAC之间的三元复合物结构揭示了相互作用的可塑性, 其中不同的接头可导致连接酶-靶标界面的不同,靶标特异性排列(图2c)。 类似地,对PROTAC施加的VHL和p38亚型可及的接口的限制,其连接子长度或特异性连接点的减少会导致p38α29的亚型选择性降解。 最近报道了尝试通过对接模拟来预测蛋白质界面的方法,但此时PROTAC的发现仍是经验性的。
与PROTAC不同,结合CRBN的IMiD缺少靶向底物的化学部分。相反,靶标通过β-发夹与CRBN结合的邻苯二甲酰亚胺基团直接相互作用,该β-发夹在所有可用的复杂结构中结构保守,其中关键位置的甘氨酸以邻位邻靠邻苯二甲酰亚胺,而该结合姿势将与任何其他不相容氨基酸(图2d)。这种独特的结构排列保留在无关的IMiD靶标中,例如锌指蛋白IKFZ1和ZNF692,激酶CK1α47和GTP结合蛋白GSPT1,并用于鉴定被沙利度胺(thalidomide)类似物降解的新型锌指蛋白。重要的是,在异双功能分子的背景下,这种相互作用似乎也得以保留。例如,靶向BTK酪氨酸激酶的CRBN募集,基于IMiD的PROTAC还会降解结合邻苯二甲酰亚胺的新底物IKFZ1和IKFZ3,从而对套细胞淋巴瘤产生协同作用和有益作用。这种双重活性增加了一些基于IMiD的PROTAC可能易于降解脱靶的特征,这些脱靶具有含甘氨酸的β-发夹degron,包括150多个锌指蛋白,这些蛋白可能会掩盖对PROTAC的表型反应的解释。治疗或影响候选药物的毒性。
目前,人类蛋白质组中600多种中的E3连接酶(CRBN,VHL,IAP,MDM2,DCAF15,DCAF16,RNF114)中只有不到10种被降解诱导小分子利用。 将配体库扩展到具有各种结构特性以及多样的时空表达谱的E3连接酶,将大大扩展PROTAC在化学生物学中的潜在应用,并拓宽未来药物开发工作的视野。 有了这个机会,我们现在总结人类E3连接酶的分类,它们在癌症中的表达谱和必要性,然后系统地分析其配体性。
三、扩展工具箱
3.1 人类E3连接酶的分类
E3连接酶可以用单遍在蛋白或聚合遍在蛋白链标记底物蛋白,其中连续的遍在蛋白分子通过不同的异肽键连接。 这些独特的多聚泛素链中只有一些,例如通过赖氨酸48分支的,会导致标记蛋白的蛋白酶体降解。 泛素化的非蛋白水解功能包括DNA修复或亚细胞定位。 对于许多E3连接酶来说,安装的聚泛素链的类型仍然是未知的,这些酶与UPS的联系及其对PROTAC的适应性也是如此。 根据可从UniProt和Reactome Pathway数据库获得的E3连接酶及其结合伴侣的注释,目前认为632个或更多人类E3连接酶中约有270个与UPS有关。
E3连接酶根据其作用机理通常分为两大类:
- HECT域酶在将泛素转移到其底物之前与泛素形成硫酯键
- RING E3连接酶通过其RING域募集E2-泛素缀合物并催化E3连接酶。直接将泛素从E2酶转移至底物。
底物结合域和RING域可以属于同一蛋白,也可以是多亚基复合物(例如CRL)的不同组成部分,其中Cullin充当底物靶向亚基和E2结合蛋白之间的蛋白质连接子(图。 2a,b)。例如,DCAF E3连接酶通常包含底物结合WD40重复(WDR)模块和介导通过衔接蛋白DDB1与Cullin 4结合的独特域。 Cullin 4同时结合RING域蛋白RBX1,后者又募集E2-泛素结合物用于后续的底物泛素化。在后期促进复合物(APC)连接酶中观察到了Cullin主题的变化,其中多亚基复合物的不同实体将E2-泛素结合物和蛋白质底物紧密相连。环之间的环(RBR ,RING-between-RING)E3连接酶是通过RING域结合E2但形成中间硫酯键(如HECT酶)的机制杂合体,然后将泛素转移至底物。
3.2 E3连接酶的表达 Expression of E3 ligases
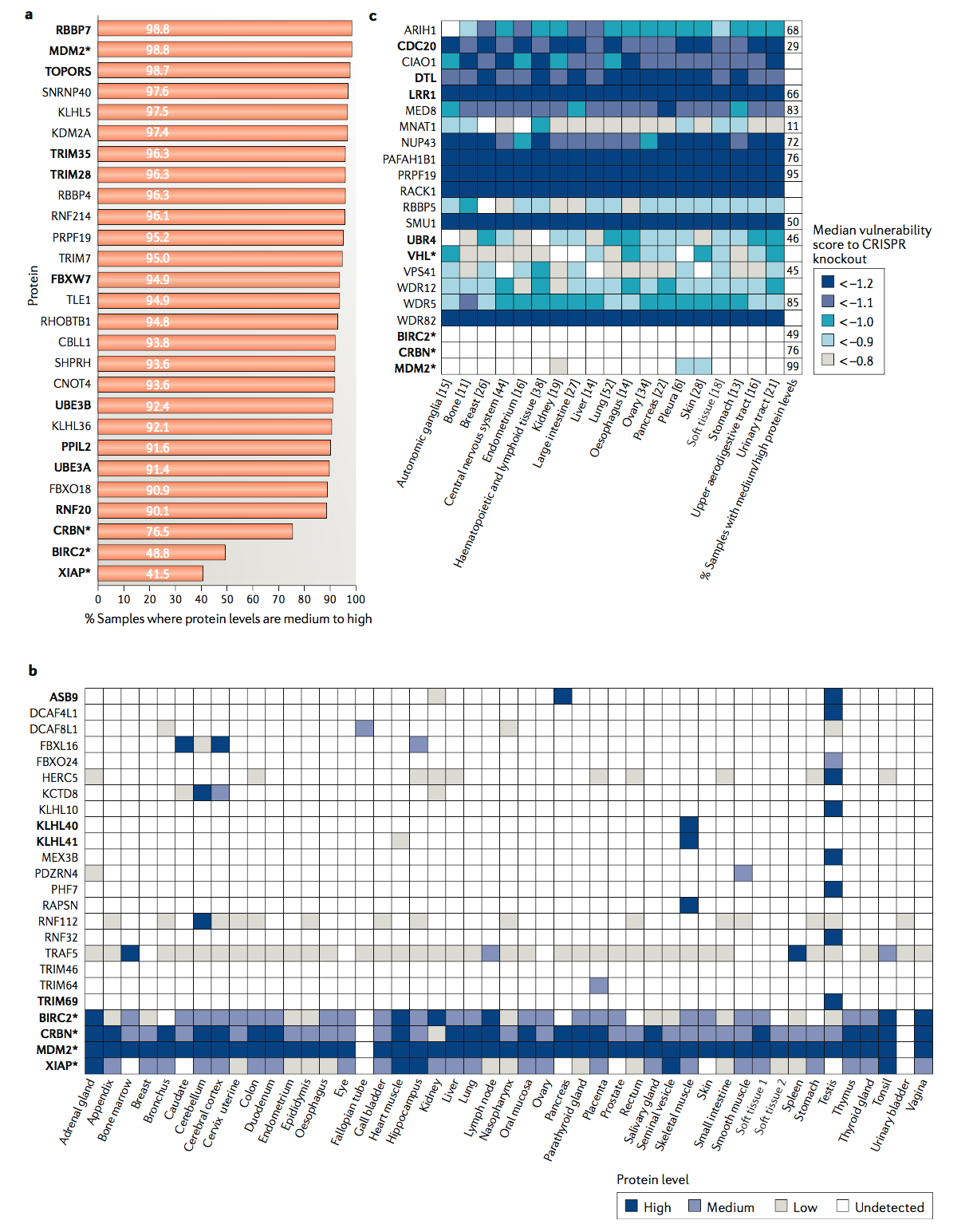
仅当所募集的E3连接酶在目标细胞或组织中可用时,PROTAC才具有活性。因此,依赖于普遍表达的E3连接酶的PROTAC可以在广泛的细胞系统中用作化学生物学工具。根据可从人类蛋白质图集(参见相关链接)获得的81种细胞和组织类型的蛋白质组学数据,在至少90%的测试细胞和组织类型中存在24种E3连接酶(图3a)。其中,UPS涉及十个(RBBP7,MDM2,TOPORS,TRIM35,TRIM28,FBXW7,UBE3B,PPIL2,UBE3A和RNF20)。 MDM2是现有PROTAC开发的一种E3连接酶,在99%的测试样品中表达,表明在各种细胞环境中,招募MDM2的PROTAC应该是有效的化学生物学工具。最近显示,基于MDM2的PROTAC可以通过降解新底物和通过与内源底物p53竞争和稳定来协同作用的催化机制和基于占用的效应(reF.60 )。这种协同作用对于药物可能是理想的,但可能使与新底物降解相关的表型模糊。预期这种效应会根据细胞中E3,PROTAC和底物之间的化学计量平衡而变化,并且可以通过将PROTAC浓度降低至竞争性差但仍具有催化活性的亚化学计量水平来降低,或者通过利用E3连接酶的非功能性结构域。凋亡抑制剂(IAP)蛋白BIRC2和XIAP被当前的PROTAC(也称为特异性和非遗传的IAP依赖性蛋白擦除剂(SNIPER))劫持,但在超过50%的细胞和组织类型中,其水平未达到可检测水平人类蛋白质图谱(图3a),可能是化学基因组学方法的责任,但对翻译研究是有利的。

募集具有组织选择性表达特征的E3连接酶的PROTAC有望为治疗应用提供独特的机会,因为它们不应降解未表达E3连接酶的组织中的靶蛋白。
根据人类蛋白质图谱(图3b),二十种E3连接酶在整个人类组织中的表达范围都很窄。 其中,已知有四种诱导其底物的蛋白酶体降解(ASB9,KLHL10,KLHL41和TRIM69)。 例如,SOCS盒E3连接酶ASB9仅在胰腺和睾丸中表达,而F-盒E3连接酶FBXL16特别在尾状和大脑皮层中表达。 与这些E3连接酶之一具有足够效力和特异性的化学手柄可以与多种底物靶向配体相连,从而使疾病相关基因的组织特异性沉默。
当前开发的E3连接酶的表达谱相对普遍,这可能会转化为招募这些酶的PROTAC产生不良影响的情况(图3b)。 这些蛋白质组学数据取决于用于蛋白质检测的抗体的质量和选择性,因此需要进一步验证,但是它们仍然说明了利用具有多种组织表达特征的E3连接酶的价值。 在此主题的有趣变化中,PROTAC可用于诱导特定细胞区室中的底物降解:最近显示,共价募集核E3连接酶DCAF16的PROTAC仅能降解核靶标。
3.3 E3连接酶在癌症中的重要性 Essentiality of E3 ligases in cancer
根据癌症依赖性图谱( depmap.org ),该图谱提供了从超过340个癌细胞系和多种癌症类型的CRISPR基因敲除研究获得的基因重要性,许多E3连接酶是必不可少的-因此可用于蛋白酶体靶向应用-在大多数癌症类型中(图3c)。特别令人感兴趣的是,APC的底物结合亚基CDC20可以诱导靶蛋白降解,并且在所有测试的癌症类型中都是必不可少的,但据人类报道,其在70%的非癌细胞中的蛋白水平低至未检测到蛋白质图谱。此外,先前已报道了一种弱的小分子配体,它与CDC20的底物结合结构域结合,这表明该结构域在化学上易于处理。针对CDC20的更有效的化学处理方法将是开发针对多种癌症类型中癌基因的PROTAC的有吸引力的工具。具有CDC20的底物竞争者阻断有丝分裂退出并诱导肿瘤细胞死亡,这可能与PROTAC驱动的致癌新底物降解协同作用。 E3连接酶复合物的基因组改变是癌细胞对VHL或CRBN募集的PROTACs的长期治疗产生的耐药机制。开发对癌细胞存活至关重要的E3连接酶是避免这种耐药机制的一种有前途的策略。
3.4 E3连接酶的可连接性 Ligandability of E3 ligases
扩展PROTAC开发的E3连接酶的库是一个引人入胜的前景,并得到以下观察结果的支持:代表不同酶和结构类别的6种E3连接酶中有5种在融合到人工配体结合域时适合于降解靶标。 但是,只有在目标E3连接酶的结构具有具有允许小分子配体结合的几何和物理化学特性的口袋或缝隙的情况下,才能实现这一愿景。 因此,基于可用的结构和化学数据,其余观点将重点放在主要类别的E3连接酶的配体能力上。
3.4.1 DCAF E3连接酶
DCAF E3连接酶是一个约有60种酶的亚家族,其中52种含有WDR结构域(图4a)。 DCAF蛋白CRBN不具有WDR结构域,是PROTAC发现中最有特征的E3连接酶之一,CRBN的晶体结构与PROTAC和非天然底物BRD4形成复合物,其中化合物和靶蛋白分别结合连接酶的非典型CULT和Lon结构域。 最近显示,一类磺胺药利用含WDR的DCAF15化学诱导癌症靶标RBM39的泛素化和蛋白酶体降解(reFs65,66)。 磺酰胺靶向的DCAF15的区域尚未确定,但WDR域(一种常见的蛋白质相互作用支架,也是该蛋白质的唯一预测域)是一个明显的候选对象。与VPR复合的同源DCAF1的晶体结构表明底物识别依赖于WDR域,这是一种类似于甜甜圈的蛋白质相互作用模块,在其他蛋白质的情况下,已经成功地被小分子配体靶向(图4a ):EED是多梳抑制复合物2(PRC2)的关键组成部分,其行为相当于DCAF E3连接酶,可募集PRC1用于组蛋白底物的泛素化,而目前在临床开发中的纳摩尔配体结合了EED WDR结构域的中心腔。高效的选择性化合物也靶向另一种WDR DCAF E3连接酶WDR5的中央腔。最近,PROTAC被证明可通过DCAF16的半胱氨酸定向共价募集来诱导非天然底物的降解。确实,与E3连接酶的共价结合不应阻止单个E3-PROTAC实体对多个底物分子的连续泛素化作用,因此有望保留PROTAC介导的底物降解的亚化学计量催化性质。
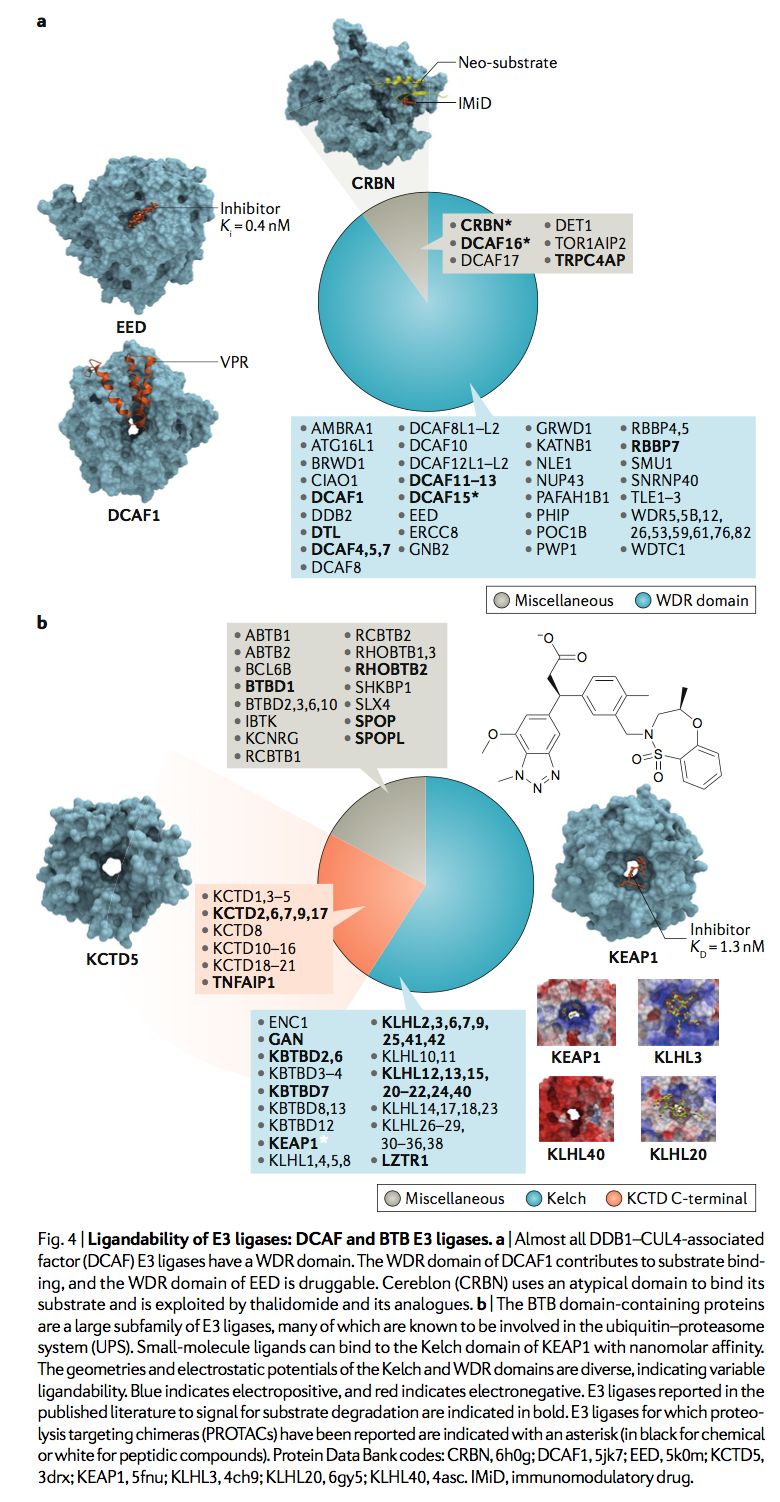
WDR域的中心腔通常较深且封闭,这是化学手柄有效结合的两个重要属性,但该位点有时可以带正电荷或带负电荷,从而降低了其配位能力。例如,与WDR5,EED,DCAF1,PAFAH1B1,ATG16L1或ERCC8相比,DDB2和RBBP4的中心腔分别是高酸性和碱性的。侧链的可塑性也可以极大地影响中心腔的配位性。例如,该位点浅并且在EED的脱辅基结构中看起来是不可配体的,并且构象重塑对于配体结合是必需的。不认为WDR5和EED会诱导蛋白酶体降解,因此不适合PROTAC发现,但是在52个含有WDR结构域的DCAF E3连接酶中,预计其中一些与UPS相关并且可以配体。 WDR结构域的外壁也可以用作蛋白质相互作用界面。例如,CDC20(一种非DCAF E3,充当APC的底物结合亚基),利用其WDR域的侧面通过其D-box基序募集APC底物,而apcin是在相同的CDC20位点可抑制D-box底物的泛素化。如下所示,在E3连接酶的多个亚家族中发现了WDR结构域和其他与结构相关的β-propeller结构,这表明这些doughnut-like结构域是底物募集和PROTAC发现的有效模块。
3.4.2 BTB E3连接酶
人们认为220种含人BTB的蛋白质中大约有90种起着Cullin 3依赖性E3连接酶的作用,并且将BTB Cullin adaptor和底物识别域独特地结合为一个蛋白质(图4b)。 含BTB的E3连接酶通常以3-box基序的存在为特征,该基团能够实现Cullin 3的高亲和力结合。大多数BTB结构域也均二聚化,为这些E3连接酶提供了两个底物识别中心,它们能够与一个CDNA内的多个杂交子结合。 单底物,例如分别由KEAP1和SPOP与底物Nrf2和Ci / Gli78的相互作用所举例说明。 劫持BTB E3连接酶的概念证明已由靶向Tau的肽PROTAC提供,可被KEAP1降解。 重要的是,该肽基于Nrf2内的一个degron位点,这表明在等效的化学PROTAC中可能不需要多价。
BTB-Kelch蛋白是E3类中最大的亚家族,对于药物开发而言似乎也是最易处理的。凯氏(Kelch)结构域折叠成一个六叶β螺旋桨,带有一个中央口袋,用于结合底物degrons或小分子。已经报道了四个BTB-Kelch家族成员的肽底物复合物的晶体结构,包括KEAP1,KLHL2,KLHL3和KLHL20。 但是,迄今为止,由于其在慢性炎性和神经退行性疾病中的治疗潜力,小分子的开发仅限于代表最典型的家族成员的KEAP183。重要的是,已经报道了KEAP1的Kelch域的低纳摩尔抑制剂,证明了该目标类别的配体能力。人类Kelch域的结构比较显示,它们的口袋形状和表面电荷存在显着变化,这可能会影响每个成员对PROTAC手柄的开发的有利程度(图4b)。通过将其亚基低聚为五聚体,在某些KCTD家族成员中也形成了独特的β-螺旋结构,如KCTD5所示,它通过泛素介导的Gβγ亚基降解来调节GPCR信号传导。在SHKBP1中也预测了WDRβ-螺旋结构域。
含BTB的E3连接酶已与多种蛋白水解和非蛋白水解泛素信号相连,这些信号可能会限制其在PROTAC中的应用或使其变得复杂。 例如,KLHL12可以诱导蓬乱的降解多聚泛素化,但它也可以与特定的辅助适配器组装在一起,以单泛素化COPII组分SEC31进行胶原蛋白运输,或诱导多巴胺受体D4.2和D4.4的非赖氨酸泛素化。 此外,KCTD家族的一些进化枝缺乏Cullin 3结合,而BTB-Kelch家族的KLHL39似乎起拮抗剂的作用,阻断KLHL20对PML和DAPK1的泛素化和降解。
3.4.3 VHL-box和SOCS-box E3连接酶
VHL-box和SOCS-box蛋白包含一个BC框,用于与衔接蛋白Elongin B和C结合;还有一个Cullin 2或Cullin 5框,分别用于组装成特定的CRL2或CRL5复合物。不幸的是,在PROTAC开发的背景下,VHL代表单例E3连接酶,因为底物结合结构域的唯一同源物VHL样蛋白(VLP)作为缺乏C末端VHL-box的优势阴性蛋白Elongin B / C和Cullin 2相互作用所需的。尽管如此,另外12种不同的蛋白质包含一个VHL-box,已证实与Cullin 2结合(图5a)。其中许多通过与新描述的C末端degrons结合来介导蛋白质破坏。这些包括两个潜在配体的Kelch域蛋白(KLHDC2和KLHDC3)。实际上,结合至C端二甘氨酸degrons的KLHDC2的晶体结构已经揭示出由三个色氨酸和三个酪氨酸残基形成的深袋。它们的低纳摩尔底物结合亲和力可能取决于掩埋的C末端羧基,后者建立了盐桥和两个氢键。因此,像BTB–Kelch蛋白KEAP1一样,这些E3口袋可能会偏爱包含酸性部分的化合物,这对细胞通透性提出了挑战。其余的VHL-box蛋白含有富含亮氨酸的重复序列(LRR1,PRAME,ZYG11B和ZER1),四三肽重复序列(APPBP2)或锚蛋白重复序列(FEM1A–C),它们的特征较少,在缺乏结构信息的情况下可能较少有利于小分子的发展。


另外37个E3连接酶使用SOCS-box域与Cullin 5形成含Elongin B / C的复合物。 众所周知,Cullin 5复合物被HIV病毒蛋白Vif101劫持,但CRL5类E3连接酶尚未被小分子靶向。 据信,超过20种人类SOCS-box蛋白可诱导其底物降解(图5a)。 尽管不存在结构数据,但在三个成员中发现了可能用于PROTAC开发的WDR域,并且据报道WSB1形成了多种泛素链类型, 除其降解VHL103之外,还包括LRRK2(reF102)的K27和K29连接泛素化(图5a)。
锚蛋白重复序列和SOCS-box家族(ASB1-18)对于某些成员的选择性组织表达谱是值得注意的。 例如,ASB9蛋白在胰腺和睾丸中特别存在(图3),而肌肉中则存在ASB11(根据RNA水平,在图3中我们没有考虑到表达水平),而ASB4过表达 在肾上腺(根据人类蛋白质图谱)和肾上腺皮质癌(根据癌症基因组图谱)。 因此,招募ASB的PROTAC具有诱人的前景,可诱导靶标的组织选择性降解。 尽管小分子配体尚未靶向锚蛋白折叠,但ASB9的结构数据显示底物结合域中并列的疏水腔,这可能至少为将来的工作提供了希望(图5a)。
也许特征最好的SOCS-box蛋白是CISH和SOCS1-7的SH2家族。 由于设计细胞可渗透的磷酸酪氨酸模拟物的困难,SH2结构域的化学易处理性历史上一直很差。 然而,如针对STAT SH2家族转录因子所发现的,SOCS3的可用肽共结构显示出扩大的疏水口袋,其可能更易于靶向。 肽共结构也可用于SPSB1-4含SPRY域的基团,包括带有抑制性环肽的例子,这些例子可能使PROTAC原理验证研究成为可能。 最后,由于至少一个成员(RAB40C)与UPS有联系,因此RAB40家族蛋白包含一个表征较差的GTPase结构域,值得进一步研究配体性。
3.4.4 F-box E3连接酶
F-box E3连接酶是约75种蛋白质的亚家族,它们使用规范的F-box结构域与衔接蛋白SKP1相互作用,介导Cullin 1结合以募集E2-泛素结合物。根据F-box连接酶的底物结合结构域的性质,它可以分为三类。 11个FBXW E3连接酶使用WDR域进行底物募集,已知其中8个涉及UPS(图5b)。其中,由PROTAC有效地招募BTRC,所述PROTAC由与BTRC相互作用的肽和METAP2,雌激素或雄激素受体的小分子配体组成,导致其各自的靶标降解。内源性靶标的磷酸二氢吡啶酮(磷酸化的肽)与FBXW E3连接酶(如BTRC和FBXW7)的中央腔结合。因此,相应的结合口袋是基本的(图5b),靶向这些位点的化学处理可能极强,这在典型的药物优化程序中可能是无法克服的挑战,但在PROTAC的背景下可以克服。实际上,PROTACS不需要有效地与E3连接酶结合(招募具有320 nM Kd亲和力的E3连接酶VHL的PROTAC在细胞中的IC50为1 nM时会降解其目标RIPK2);此外,以前的工作表明,创造性的接头化学作用可以对PROTAC的物理化学性质产生积极影响。
FBXL是另一类F-box E3连接酶,依赖富亮氨酸重复序列(LRR)进行底物结合(图5b)。 有17种FBXL,其中15种已知介导蛋白质降解。 与底物结合的FBXL3的晶体结构沿LRR结构域未显示任何明确定义的囊袋,并且目前尚不清楚用于PROTAC发现的FBXLs的化学易处理性。 F-box E3连接酶的最后一类是42个FBXO连接酶,其中至少20个与UPS相关。 FBXO连接酶可以使用多种底物结合结构域,包括在其中六个中发现的F-box-associated(FBA)域(图5b)。 FBXO44的结构研究表明,在FBA结构域的表面存在孔洞,但迄今为止尚未描述任何配体。
3.4.5 IAP E3连接酶
IAP蛋白构成一小类的五个E3连接酶,它们通过杆状病毒IAP重复(BIR)域结合底物蛋白(图5c)。与上面讨论的E3连接酶家族不同,IAP E3连接酶通过RING域直接与E2蛋白相互作用。由于其抗凋亡功能,IAPs是癌症治疗的靶标,并且开发了利用底物结合位点的小分子拮抗剂,包括非肽模拟化合物,具有低纳摩尔效价(图5c)。结合口袋在结构上是保守的,但是同源物之间存在显着的侧链多样性(图5c),最近报道了具有窄选择性分布的配体。募集用于目标降解的IAP的小分子(也称为SNIPER)是最早描述的PROTAC之一。最初的化合物依赖于IAP配体Bestatin,该配体以中等亲和力结合BIRC2,并诱导其自身泛素化和降解,从而限制了对目标底物的作用。后来从更强大的IAP拟肽模拟配体衍生的下一代PROTAC被证明可以有效地诱导多种蛋白的敲低,如ERα,BCR–ABL,BRD4或PDE4。尽管降解ERα的PROTAC与BIRC2结合更有效,但是使XIAP沉默对化合物的活性具有更明显的影响,这表明XIAP在介导靶标降解中起主要作用,可能是由于E3的相对数量或亚细胞位置连接酶和底物。 IAP E3连接酶的组织表达谱是多种多样的,BIR结构域在化学上是易处理的,未来的IAP选择性PROTAC应该是用于化学生物学或治疗应用的有价值的工具(如专利WO2017182418和WO2017211924所示)。
3.4.6 APC E3连接酶
APC是一种大型的多亚基E3连接酶,可通过靶向细胞周期蛋白B和丝氨酸蛋白进行蛋白酶体降解来诱导有丝分裂的退出。底物识别是通过CDC20的WDR域或紧密同源的FZR1 / Cdh1进行的。如上所述,CDC20在所有癌症类型中都是必不可少的,但在大多数正常组织中其表达水平却很低,这使其成为PROTAC发现的诱人E3。 CDC20 WDR结构域的结构与底物肽复杂地解析,其中经典RxxL D-box基序插入WDR域侧面的疏水腔中。结构研究表明,D-box肽可以结合FZR1的WDR域中的类似口袋。以低微摩尔亲和力结合CDC20的小分子配体apcin占据了连接酶的D-box结合位点,表明可以开发利用该位点的PROTAC .这些数据将CDC20定位在有利的组织表达谱和具有良好的化学易处理性,可用于化学诱导癌基因降解。
3.4.7 HECT E3连接酶
29种人类HECT E3连接酶的泛素连接酶活性(大多数参与了UPS)依赖于反应中间体,其中泛素链与HECT催化结构域形成硫酯键,然后转移至底物。因此,预计结合HECT结构域的化合物将充当催化抑制剂,未来的PROTAC应当改用其他结构域。例如,六个HECT E3连接酶(HERC1-HERC6)包含一个与WDR和Kelch域结构相关的环形螺旋桨状RCC1类结构域(RLD)(蛋白质数据库代码:3kci,4o2w,4l1m)(图6a)。到目前为止,还没有关于RLD的小分子配体的报道,但是中心腔很深,可能适合PROTAC的发现。九个HECT E3连接酶具有WW底物结合结构域(图6a)。与肽底物复合的ITCH和NEDD4 WW域的晶体结构揭示了一个浅但疏水的结合位点,该位点可容纳脯氨酸丰富的基序,但配体性不清楚。

3.4.8 TRIM E3连接酶
TRIM蛋白是大约73种E3连接酶的家族,它们通过规范的RING域与缀合泛素的E2蛋白直接相互作用。 其中,已知有31个参与UPS,但由于许多TRIM E3连接酶没有功能特征,因此这个数目可能会增加。 这些蛋白质通常通过中央螺旋线圈结构域同源二聚体,并通过通常为SPRY结构域的C末端模块结合其底物(图6b)。 TRIM21的SPRY结构域是自身免疫性疾病中的主要自身抗原,与免疫球蛋白IgG126的Fc区形成复合物,从而在IgG结合位点显示了一个明确定义的囊袋,可被PROTAC靶向(图6b)。 同源位点TRIM25(reF.127)中的相应位点较浅,尚不清楚SPRY域的化学易加工性(迄今为止尚未报道其配体)。
在TRIM24,TRIM28和TRIM33的C末端发现的明显可配体的结构域是溴结构域。该结构模块识别乙酰化的赖氨酸,并在最近几年出现 肿瘤学和炎症方面有希望的目标类别。实际上,小分子配体可以以低纳摩尔亲和力与TRIM24的溴结构域结合,并且晶体结构表明该抑制剂深深地插入了溴结构域129的乙酰赖氨酸结合口袋中(图6b)。这三种泛素连接酶均参与UPS,TRIM24可以结合肿瘤抑制因子p53的乙酰化肽,导致泛素化和靶标降解。在这方面,衍生自现有TRIM24配体的PROTAC可通过催化机制同时诱导新型底物的降解,并通过基于经典占用的竞争机制稳定内源性底物p53。相反,最近将一个溴结构域配体与VHL募集的化学手柄相连,以降解TRIM24(reF.27)。 TRIM28被广泛表达(图3),并且是开发适用于多种细胞系统的蛋白酶体靶向化学生物学工具的良好候选者。
位于三个TRIM E3连接酶(TRIM2,TRIM3和TRIM32)的C末端的最后一个感兴趣的底物结合域是NHL域。 这些蛋白没有可用的NHL结构,但是对不相关蛋白的结构研究表明NHL结构域采用的β-螺旋桨拓扑结构(PDB代码5ex7)与其他连接酶中发现的WDR,Kelch和RLD结构域非常相似。 可能适合将来招募参与UPS的TRIM32的PROTAC的发展(图6b)。
虽然我们在这里重点介绍E3连接酶的主要类别,但我们希望不属于明确定义组的非典型蛋白质也将被证明适合PROTAC的发现。 例如,GID4 E3连接酶复合物的亚基GID4具有底物结合结构域,具有深的,封闭的和可配体的口袋,可识别蛋白质底物的氨基末端脯氨酸残基,从而导致其蛋白酶体降解。
四、展望
展望未来,仍然存在重要的问题。哪一部分“不可负担的”蛋白质组将证明适合靶向降解,哪些靶标将导致真正的转化疗法?尽管已取得重要进展,但仍有大量工作要做。我们建议开发更多的E3连接酶“工具”将成为该领域的助推剂,因为我们开始更充分地确定支配有效靶标-连接酶配对的因素。另外,由于不同的E3连接酶在不同的时间和不同的组织中表达,因此可以通过组织特异性靶标敲低来实现附加的机会层(和复杂性)。由于E3连接酶的“工具”主要代表配体,因此我们建议直接结合测定法作为E3连接酶的有吸引力且合适的命中鉴定策略。一个机会在于DNA编码文库(DEL),它有可能在一个或多个密切相关的实验(> 109种化合物)中取样巨大的化学空间。备选地,基于片段的方法提供了筛选选自化学上多样且有吸引力的低分子量药效团的更有限的化合物组的机会。在这两种情况下,我们都相信,基于结构的设计和分子建模的应用将极大地帮助从最初的开发到最优化的工具的发展。
尽管E3连接酶的大小,广度和多样性提出了相当大的挑战,但它们同样提供了巨大的机会。 确实,考虑到仅利用少数几种连接酶的化合物所取得的进展,有趣的是,当我们从这个基因家族的表面抓下时,了解蛋白质降解的未来将如何发展。
参考资料
- Matthieu Schapira et al. Targeted protein degradation: expanding the toolbox. Nature Reviews Drug Discovery (2019).
