【5.3】基于结构的药物设计
一、SBDD概述
基于结构的药物设计 Structure Based Drug Design (SBDD)
基本思想:是根据配体(ligand),即外源性小分子药物或内源性活性物资,与生物 体内的靶点(target)相互作用产生生物活性,从而治疗靶点相关的疾病。
基本原理:基于受体与配体的“锁-钥”原理,即配体像一把钥匙,通过形状、化学 性质等互补匹配受体活性位点的锁芯,从而使受体-配体紧密结合
分类
- 基于受体结构的药物设计(receptor structure-based drug design)
- 基于配体结构药物设计(ligand structure-based drug design)。
前者称为直接设计(direct design)方法,后者称为间接设计(indirect design) 方法

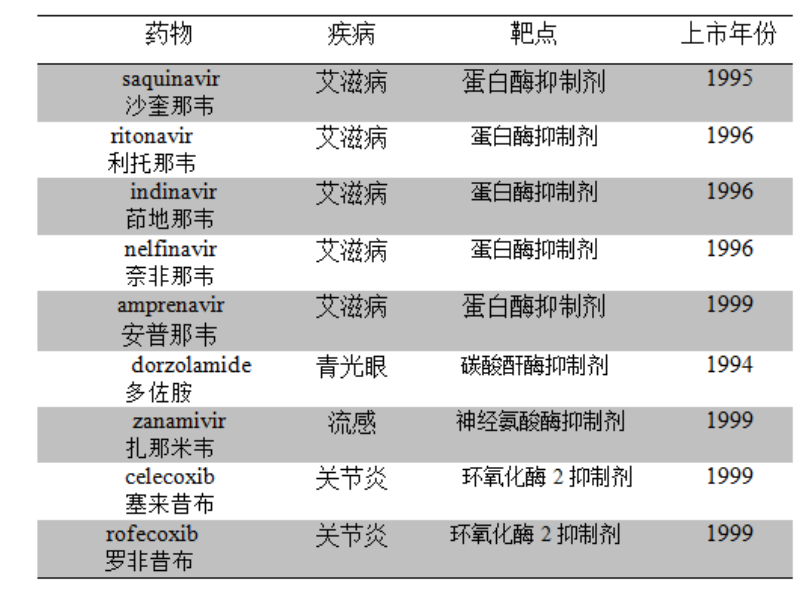
源于SBDD的治疗药物
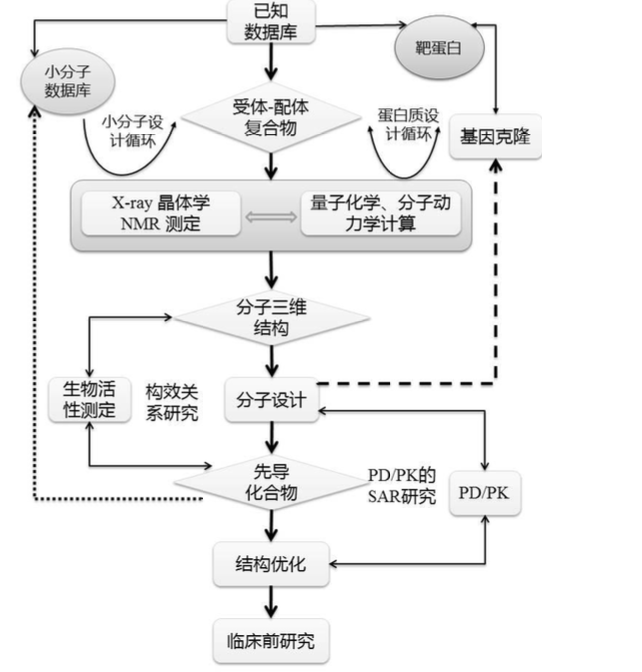
计算机辅助药物设计 (computer-aided drug design, CADD)
- 应用各种理论技术方法和分子模拟技术将SBDD的思路以计算机方法加以实 现,为药物设计提供了强有力的基本工具和手段
- CADD是多学科的有机结合和综合运用
CADD基本策略与流程
二、基本理论、技术和方法
2.1 药物受体相互作用理论——受体理论
一种有效的药物必须符合以下两个要求:
- 与机体内的某一种或多种分子靶点发生相互作用(药效学要求)
- 到达靶点(药动学要求)
基本概念:
- 受体:能识别和结合生物活性物质,并产生生物效应
- 内源性活性调节物与受体的相互作用是维持机体机能的基本生理学机制
- 外源性药物可以作用在受体而干预生理生化作用
- 受体激动剂(agonist):启动了受体的生物学功能,如一些内源性物质
- 受体拮抗剂(antagonist):药物与受体结合后阻碍了内源性物质与受体 结合,而导致生物作用的抑制
2.2 药物-受体相互作用方式
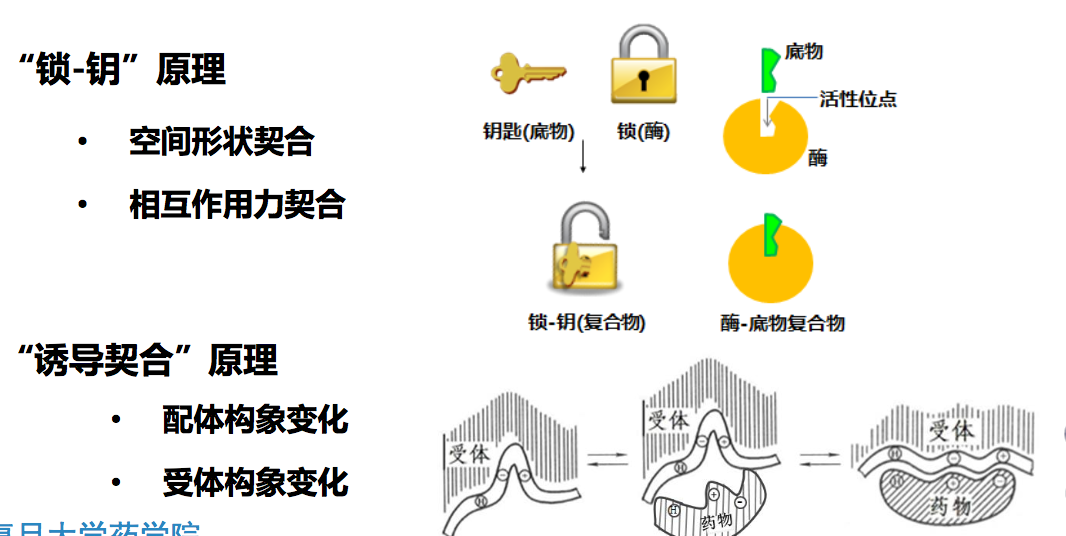
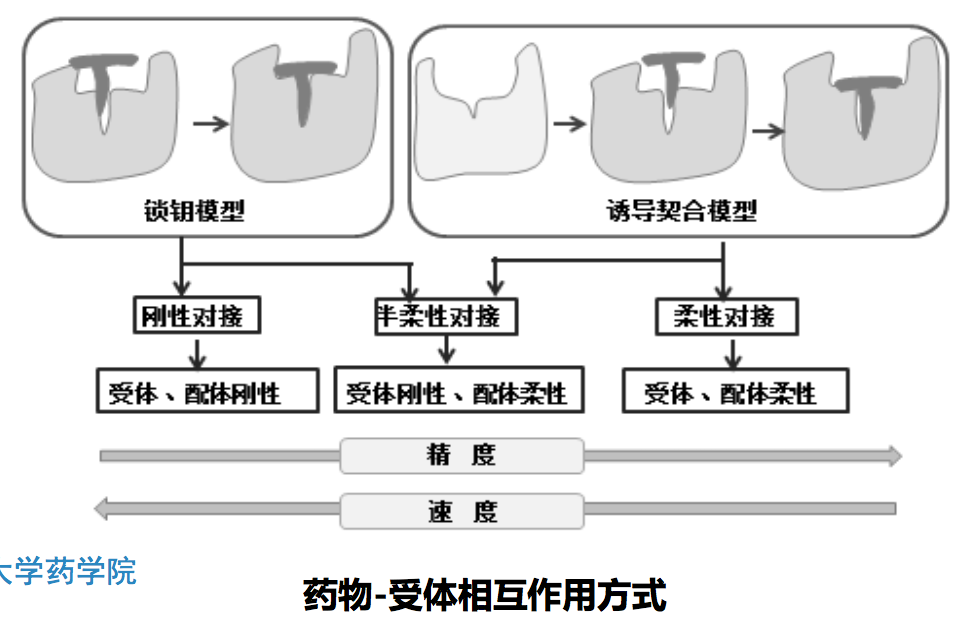
分子识别 (molecular recognition):生物分子之间发生特殊的、专一性的 相互作用,通过分子间相互作用力(interaction force)而结合

2.3 药物-受体相互作用力的类型和性质
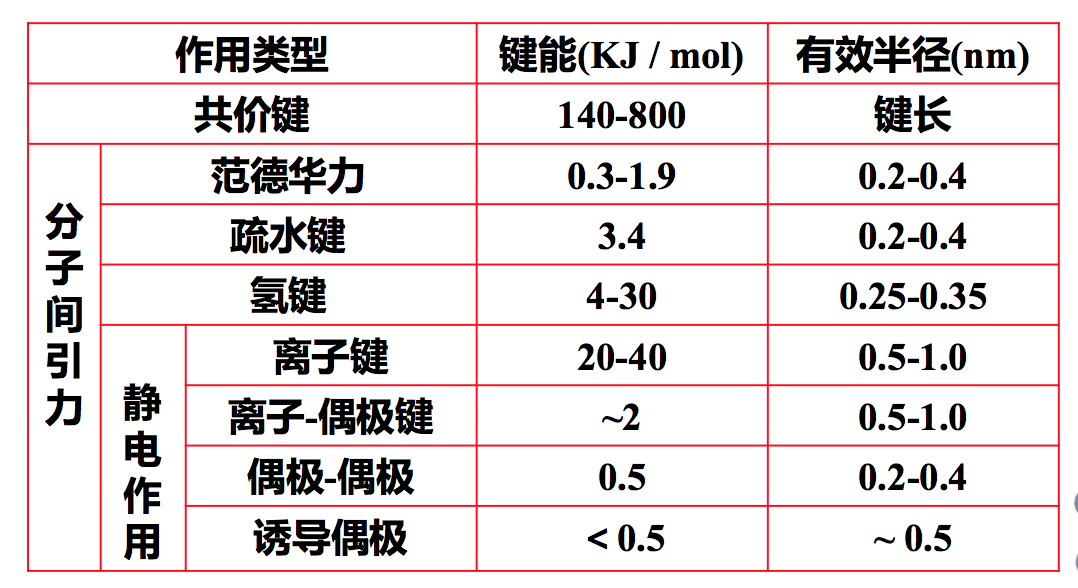
药物-靶点相互作用力的类型
- 共价键(covalent bond):
- 键能高,约140~800 KJ /mol,结合牢,不可逆;高活性,缺乏特异选择性,驻留时间较长,治疗指数高,能克服耐药性
- 范德华力(van der Waals Force, VDW)
- 相邻的电中性原子之间存在的微弱的吸引力,它由瞬间偶极引起,极性分子和非极性分子都存在着这种力
- 主要的分子间作用力
- 原子间距离为0.3~0.5 nm,范德华力的能量 为1.9 KJ / mol
- 疏水键(hydrophobic bond):
- 两个不溶于水的分子间的相互作用
- 氢键(hydrogen bonding)
- 重要的生物大分子-药物作用方式
- 氢原子与负电性杂原子共价结合后,与另一具有未共用电子对的杂原子形成一种弱的静电引力,键能约5 KJ / mol。
- 形成氢键需要有氢键供体(hydrogen bond donor, HD)和氢键受体(hydrogen bond acceptor, HA)
- 氢键相互作用 (X, Y一般为N, O, F )
- 静电作用(electrostatic interaction)
- 离子键(ionic bonding)
- 偶极-偶极(dipole-dipole interaction)
- 荷转移作用(charge transfer interaction)
- 通过供体分子的电子从最高占据分子轨道(HOMO)转移到靶点分子的最低空缺分子轨道(LUMO)
- 螯合作用(chelation)
- 是由含有二个或二个以上供电子基团(配体)的化合物(通称螯合剂, chelator) 与金属离子通过离子键、共价键或配价键相连接,形成环状结构的螯合物的过 程
- 二齿配位体(bidentate);三齿(tridentate)或多齿配位体(polydentate)
三、分子三维结构的理论计算方法
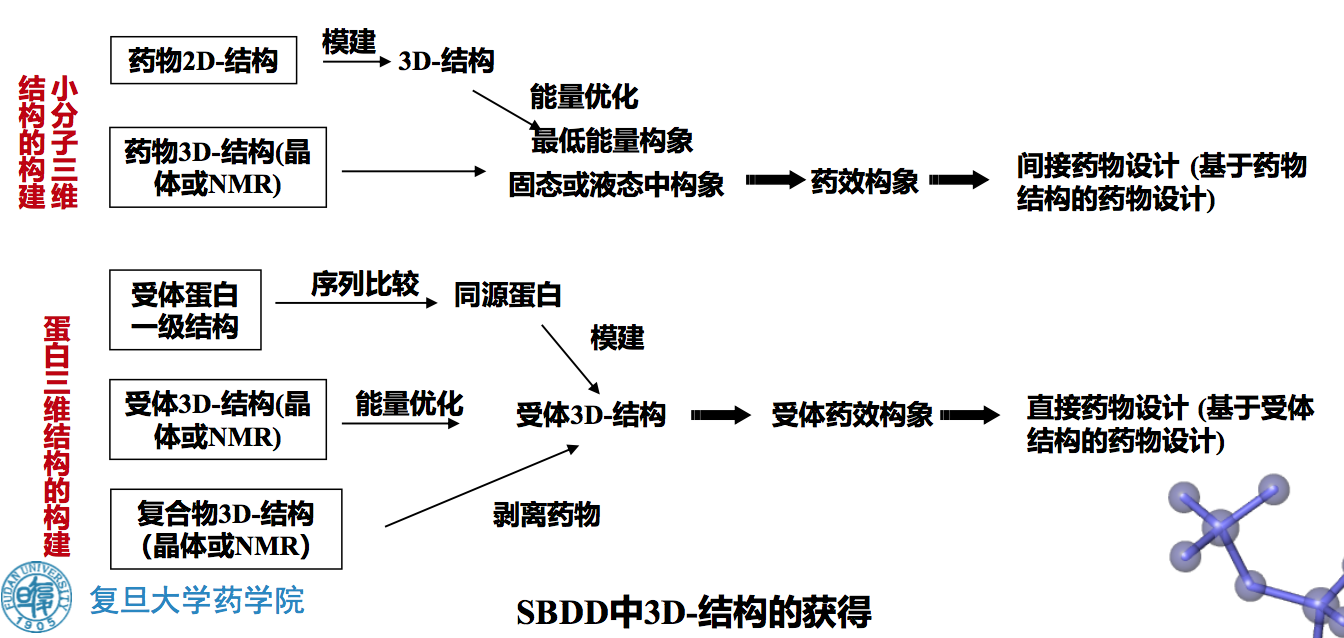

3.1 量子化学
- 量子论
- 原子中的电子只能处于包含基态在内的定态上,电子在两个定态间跃迁改 变它的能量,同时辐射出一定波长的光,光的波长取决于定态之间的能量差。
- 量子力学
- 研究微观粒子运动规律的理论。用波函数描写粒子的运动状态,以薜定谔 方程 确定波函数的变化规律,并对各物理量进行计算。
- 量子化学
- 以量子力学的基本原理和方法来研究化学问题的学科。 可计算分子的各种参数,如分子结构、电子结构、系统总能量和各个轨道信息。
3.1.1常用的方法
从头计算(ab initio methods):
- 以基本物理常数以及元素的原子序数,不借助于任何经验参数,求解薜定谔 方程 (Schrōdinger Equation): Ĥψ (r) = E ψ (r)
- 计算结果精度高,可靠性大,但是计算量极大,消耗计算机时太多 • 从头计算法的软件有Gaussian,SPARTAN等
密度泛函方法(Density Function Theory, DFT):
用电子密度取代波函数做为研究的基本量,其将电子能量分为几个部分,动能、 电子-核相互作用、库仑排斥,以及其余部分的交换相关项,所有项只是电子密 度的函数。
半经验计算法(semiempirical methods)
- 采用实验值拟合的经验参数,大大提高计算速度,计算精度较差
- 半经验计算法的软件有:MOPAC和AMPAC等
3.1.2 量子化学计算的优缺点
- 可计算出分子的理化参数、分子结构的电子结构,几何构型和易与亲电试剂或 亲核试剂反应的部位
- 只适用于计算分子量较小分子,计算时间长
3.2 分子力学(molecular mechanics) 、力场方法(force field method)
- 基于经典牛顿力学方程的一种计算分子的平衡结构和能量的方 法,来研究分子体系的结构和性质
- 计算量小,分子力学可研究包括成千上万个原子的分子体系, 包括有机小分子、生物大分子
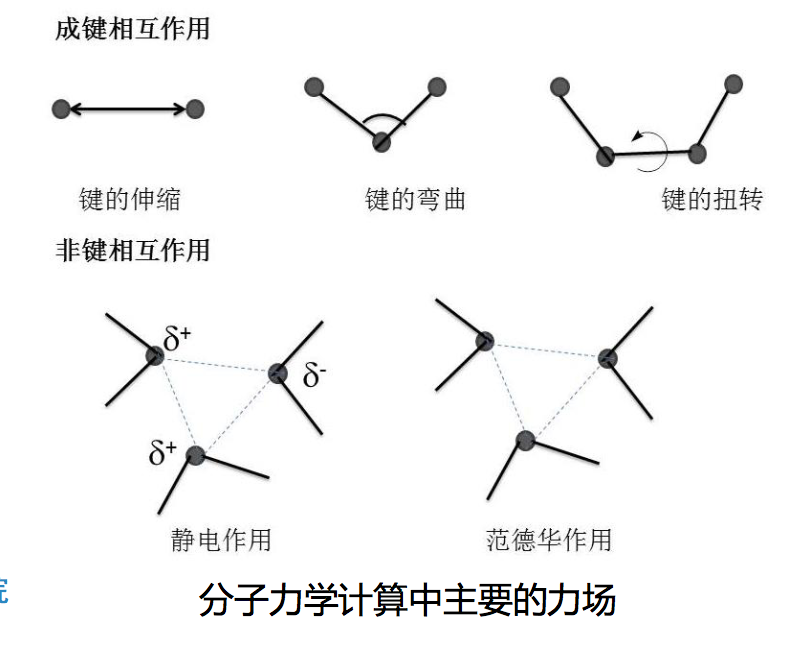
- 力场总能量,即分子总能量 = 键合作用 + 非键作用
分子力场(Force Field): 分子力学计算时采用的一套参数和方程进行体系总能量计算

- 成键相互作用(bonded interaction)
- 非键相互作用(non-bonded interaction)
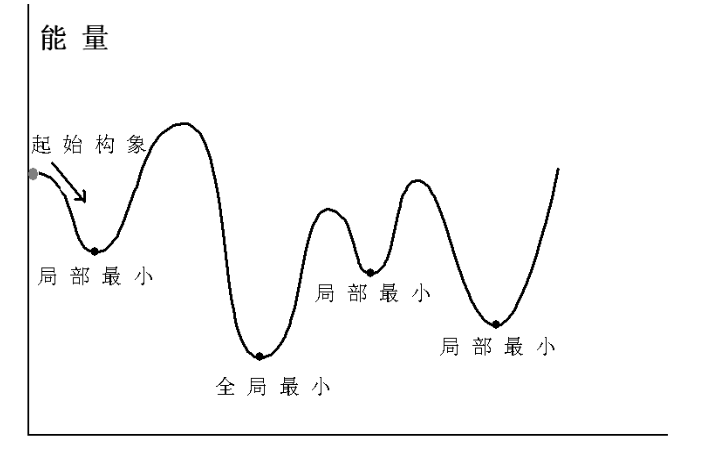
为了确定分子低能构象,就需要求解与其相对应的分子势能面上的极小值,这 一过程称为能量优化(energy minimization)
分子力学能量优化算法
- 最陡下降法(SD)
- 共轭梯度法(CG)
- 牛顿-拉普森法(Newton-Raphson)
- BFGS(Broyden、Flecher、Goldfarb和Shanno)方法
分子力学计算的优缺点
有点:
- 可计算出分子的总能量
- 计算速度快
缺点:
- 计算优势构象时易于陷于局部最小的能量
- 不能计算与化学键形成和断裂相关的体系
3.3 QM/MM
- 将量子力学方法(QM)的准确性和分子力场方法(MM)的高效性有机地结合在一起对体系进行不同尺度的计算;
- 该方法基本思想是用精确的量子力学处理感兴趣的化学反应中心,如酶和底物的结合位点,该部位划分为QM区域,其余部分如酶以及其它溶液环境区域用经典分子力学来处理,划分为MM区域
3.4 分子动力学(molecular dynamics)
- 分子动力学是研究分子构象及其它性质随时间变 化的重要工具。
- 分子动力学是在分子力学的基础上描述分子运动 随时间演化的方法
- 分子动力学在由分子体系的不同状态构成的系综 中抽取样本,从而计算体系的构象和能量,并以 此为基础计算体系的热力学量和其他宏观性质
分子动力学计算的优缺点
优点:
可给出体系的动态信息,用于研究生物大分子结构和功能的关系, 给出药物与靶点作用的动态信息。
缺点:
计算结构的准确性取决于选择正确的力场和参数值
四、分子模型技术
- computer aided molecular modeling, CAMM 计算机分子模型技术
- computerized molecular modeling, CMM 分子模型技术
- molecular modeling,MM 分子模拟
利用计算机图形学进行分子模拟的技术
为三维结构研究的有效手段——利用计算机来构造、显示、分析和贮存复杂的分子模型,提供直观的分子立体形象,观察分子间的相互作用
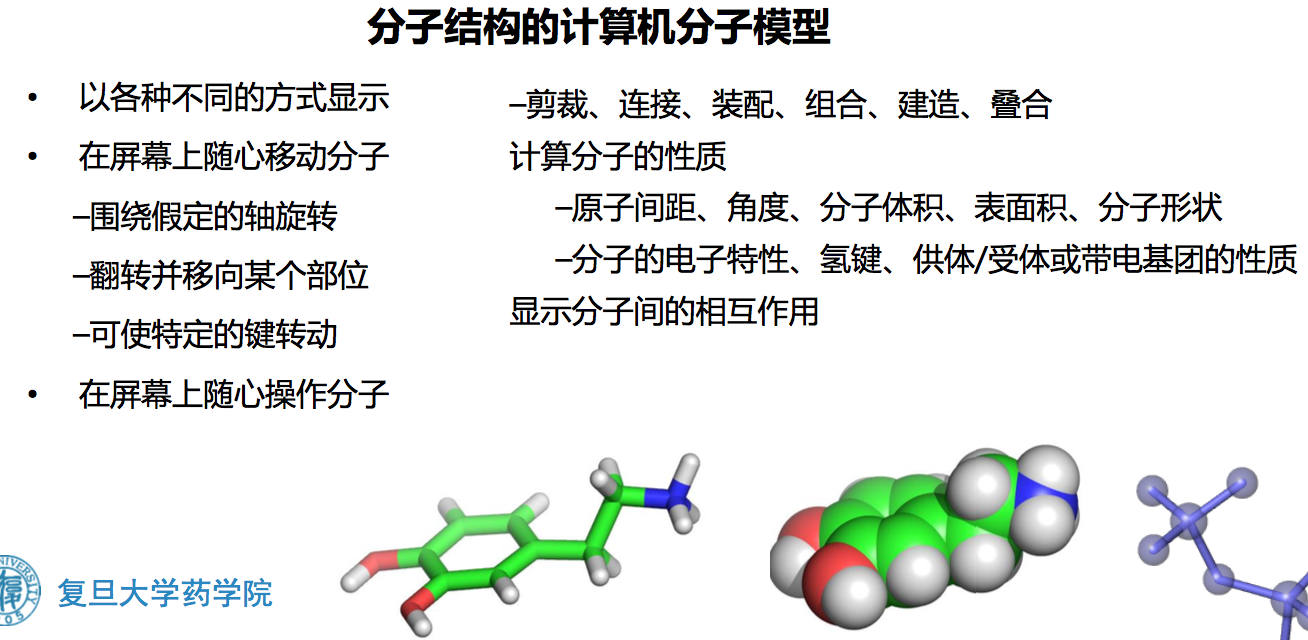
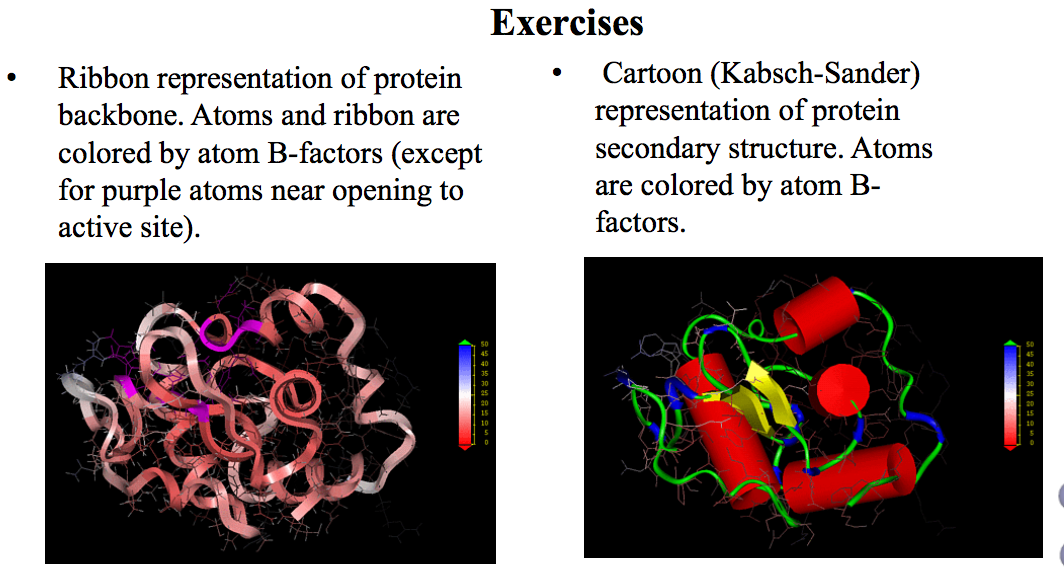
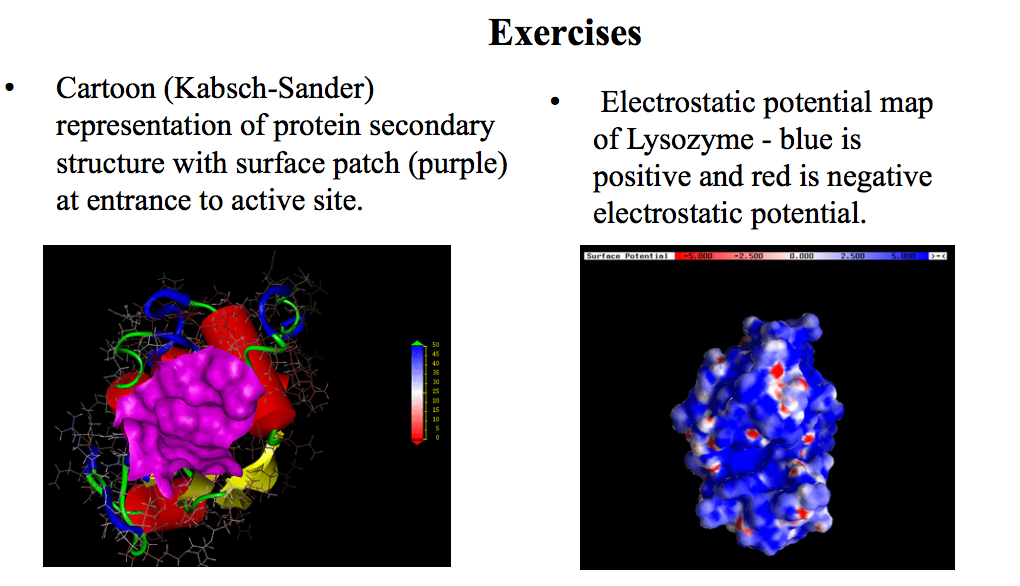
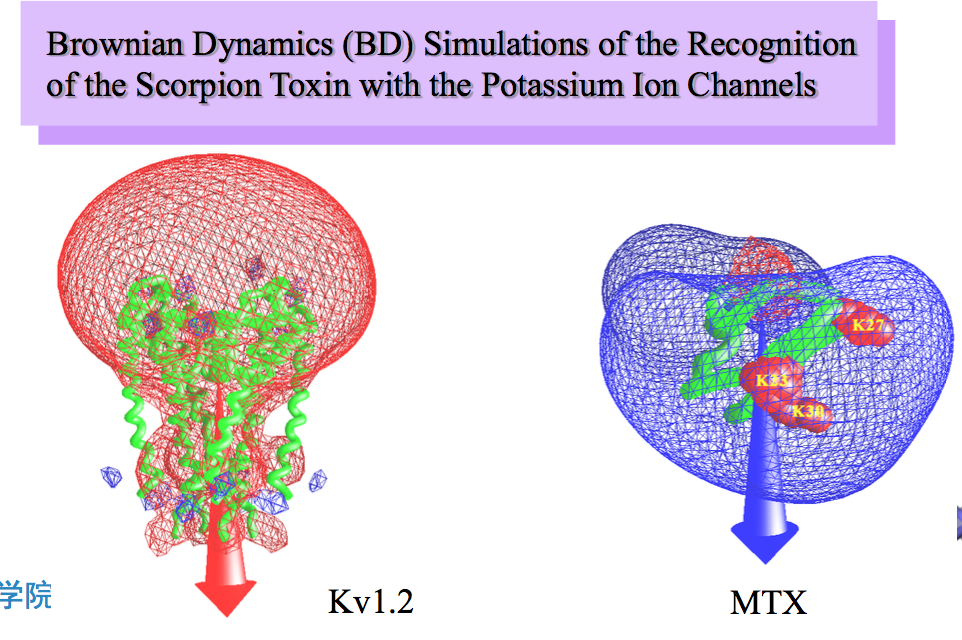
分子三维结构的显示方式

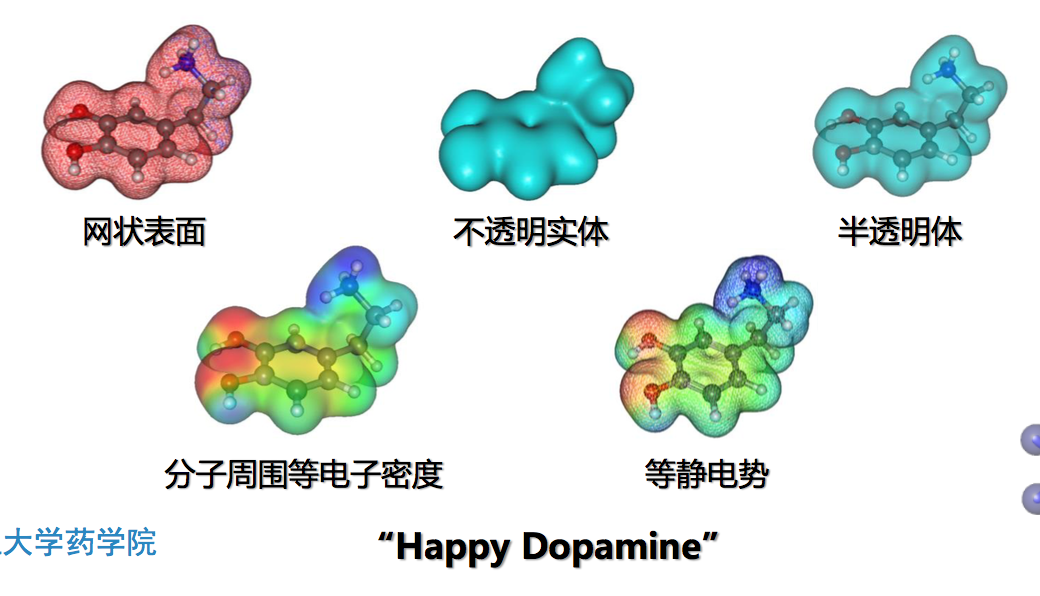
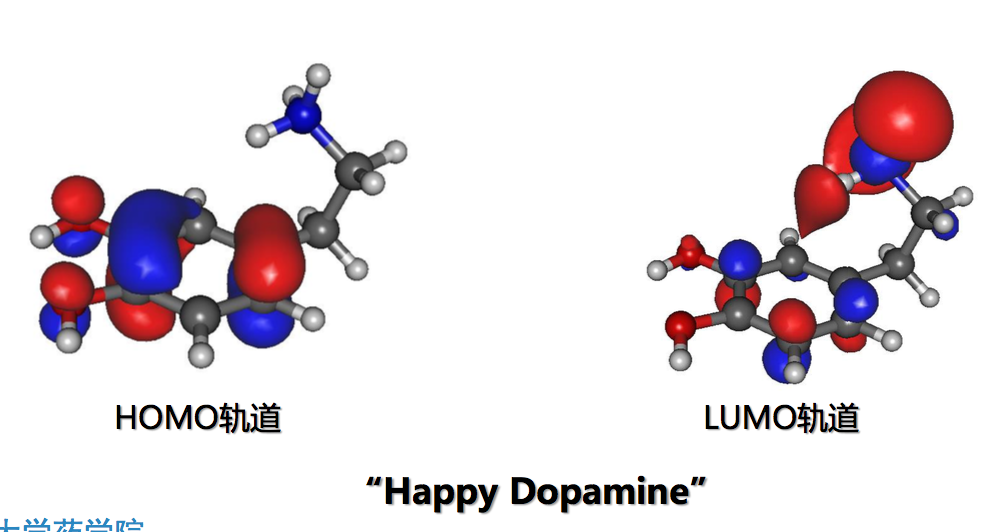
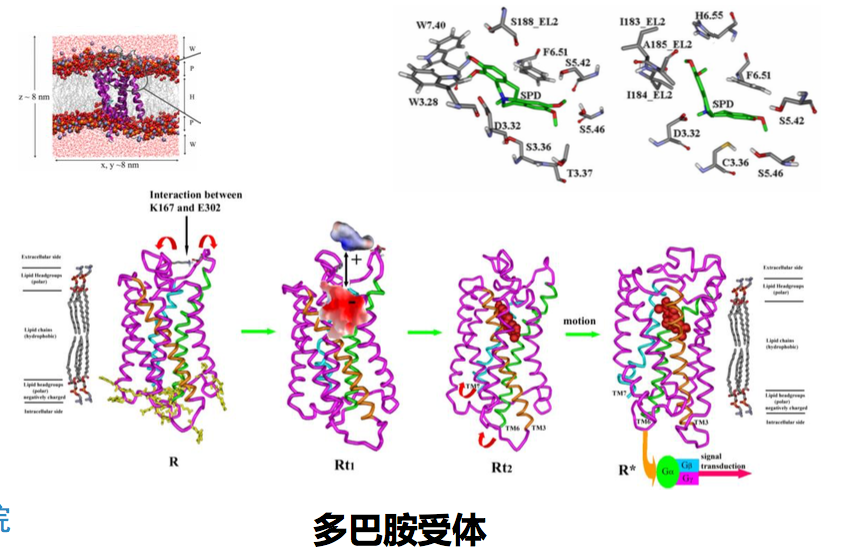
生物大分子结构的表示方式



参考资料
- 复旦大学 李伟 老师的 《药物设计学》 课件
