【5.2.2】靶向“不可成药蛋白”
不可药物靶点是指具有:
- 缺乏明确的配体结合口袋
- 非催化的蛋白‑蛋白相互作用结合区域
- 蛋白晶体结构研究极少
且通过传统方法“难以成药”或“尚未成药”的但具有临床意义的治疗靶点。
本综述介绍了RAS 和其他“不可成药”的靶点,并从靶向不可成药靶点方法的创新和新颖的药物发现技术两个方面进行介绍。
一、“不可成药蛋白”
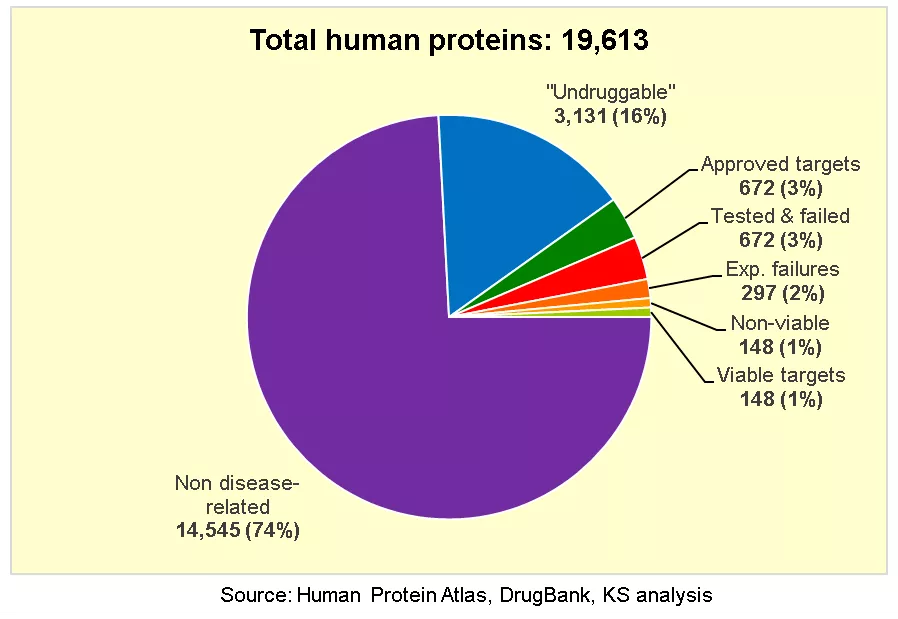
药靶是在疾病发展中起重要作用的生物大分子,可以通过外源分子治疗来治愈疾病。在过去的几十年里,在药靶的发现方面取得了巨大的突破,对疾病机制的研究促进了数百个已成功的药靶的鉴定,包括激酶、 受体和通道蛋白。激酶是代表性的“可成药”靶点, 因为它们满足以下可成药性条件:
- 可成药靶点与疾病的进展密切相关,激酶是致癌蛋白,它们的突变或过度表达会导致肿瘤发生。
- 可成药靶标通常包含明确定义的疏水口袋, 可实现高亲和力和特异性配体结合。
- 口袋中配体的结合改变了可成药靶蛋白的活性或功能(图1)【1】。
对于激酶,抑制剂可以结合在 ATP 袋、相邻的疏水袋或变构袋上以抑制激酶活性,从而引发下游变化:底物磷酸化降低、细胞信号通路下调、细胞生长受到抑制。
可成药靶点涵盖了广泛的疾病相关酶,然而,它们仅占整个人类蛋白质组的一小部分 (~2%)。
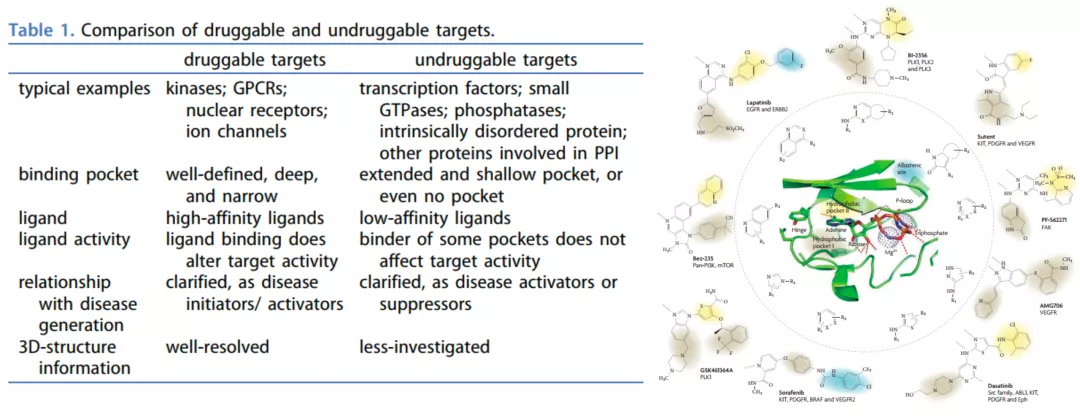
图1 药靶和不可成药靶点的比较以及多种激酶抑制剂结构
首先,不可成药的靶点属于药物靶点,必须有强有力的证据表明针对这些目标开发药物具有临床意义。 接下来,它们具有一些难以通过传统方法进行药物设计的特点,包括:
- 具有扩展和平坦的功能界面,并且缺乏明确的配体结合口袋。
- 缺乏特定配体来使得靶蛋白发挥功能。
- 一部分不可成药的靶点是疾病抑制剂,因此,药物需要激活蛋白质活性,这使得药物开发更具挑战性。
典型的不可成药靶标有:
- 转录因子:例如 c‑myc 和 p53
- RAS家族蛋白
- 磷酸酶
- 内在无序蛋白质
- 参与 PPI 的其他蛋白质:比如抗凋亡 Bcl2 蛋白家族。
二、靶向“druggable targets”的方法
以下总结了几种常见的治疗不可成药靶点的方法(图2):

图2 多种针对不可成药靶标的方法
2.1 双功能分子
通过设计双功能分子进行特异性降解靶蛋白,蛋白水解靶向嵌合体 (PROTAC) 是一种新兴的治疗实体, 在癌症和其他疾病中显示出强大的效力,可降解基于泛素‑蛋白酶体系统的不可成药的目标蛋白 (POI) 。值得高兴的是,目前已有处于临床研究的PROTAC分子,如ARV‑110和ARV‑471。

《Targeted protein degradation: A promise for undruggable proteins》
除了泛素‑蛋白酶体系统外,研究人员还利用其他内源性降解途径来实现靶向蛋白质降解。溶酶体靶向嵌合体 (LYTAC) 利用内体‑溶酶体途径降解 POI。与靶向细胞内蛋白的 PROTAC 不同,LYTAC专注于细胞外或膜结合蛋白。自噬靶向嵌合体 (AUTAC) 是另一种靶向胞质蛋白的双功能降解剂,作为不能被蛋白酶体降解的蛋白质的补充方式。
值得注意的是,AUTAC 能够降解体积较大的靶标,包括蛋白质聚集体以及受损或功能失调的细胞器。还有分子胶降解剂作为目标特异性降解剂,促进 E3 连接酶与目标蛋白的结合,分子胶降解剂因其合适的分子量和药代动力学(PK)特性而具有广阔的应用前景(图3)。

图3 多种双功能分子降解剂的比较
2.2 共价药物
共价药物自阿司匹林和青霉素的偶然发现以来历史悠久,但曾因毒性和脱靶效应而并未受到重视。 然而, 最近二十年见证了共价药物的复兴,靶向共价抑制剂(TCIs)主要通过两步发挥功能:
- 化合物首先与配体口袋非共价结合,将共价弹头放置在目标残基附近
- 共价弹头与靶蛋白氨基酸共价结合,第二步通常是单向的、不可逆的(可逆共价抑制剂除外)
共价抑制剂具有以下优势:
- 较高的功效但更低的剂量,适用于缺乏高亲和力配体的不可成药靶点;
- 克服内源性底物的竞争
- 药代动力学性质好,能够延长疗效时间。
2.3 肽类药物
尽管小分子药物将一些不可成药的靶标转化为可成药的靶点,但受其大小的限制,肽类药物介于小分子药物和生物大分子之间,同时满足中等细胞渗透性和更大的药物‑靶点界面的要求。因此,它们非常适合针对一类不可成药的靶标(即 PPI)。
一般的小分子药物界面覆盖面积在300‑1000 Å左右,而典型的 PPI 界面跨度约为 1500–3000 Å,此外,PPI 界面的特点是缺乏疏水腔来完全容纳配体。因此,PPI抑制剂需要具有更高的分子量和更复杂的拓扑结构,超出Lipinski 的五法则。
肽模拟物/蛋白质模拟物是通过合理设计、展示技术或文库筛选产生的另一类肽衍生药物。它们可能包含非天然氨基酸构建块和环状结构, 在药理特性和化学多样性方面提供优于天然肽的优势,肽模拟物已被开发用于治疗动脉粥样硬化和炎症的细胞因子信号抑制因子 (SOSC)。
基于肽的药物的例子在不可成药的 KRAS 和 p53 的药物发现中很普遍。一种名为 cyclorasin 9A5 的细胞渗透性环肽(cell-penetrating peptide, cpp)可以与 RAS 结合并阻断其与下游效应物的相互作用。
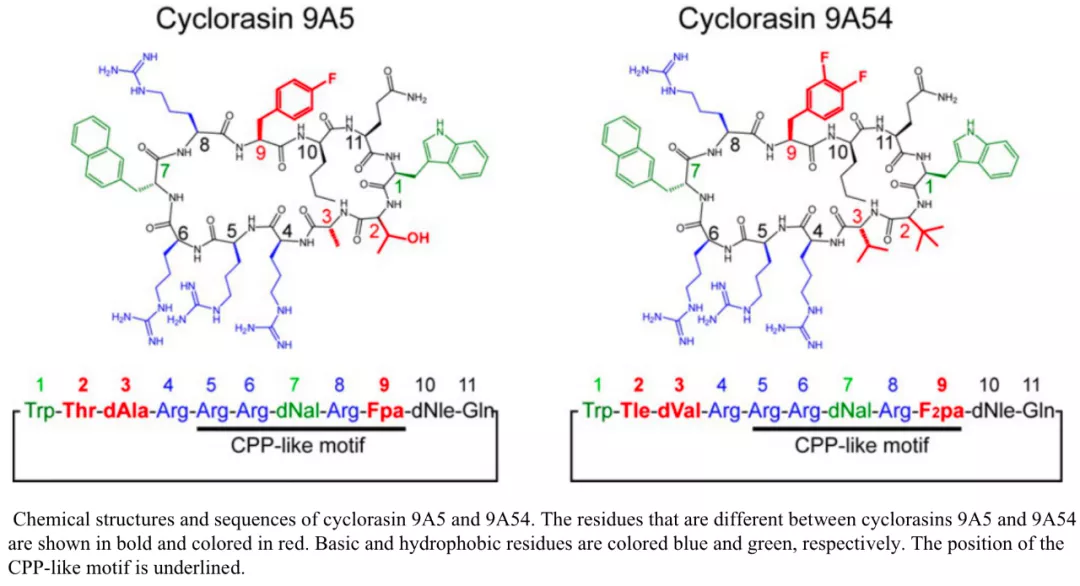
《Conformational Plasticity of Cyclic Ras-Inhibitor Peptides Defines Cell Permeabilization Activity》
2.4 蛋白类药物
基于蛋白质的药物具有高度组织化的结构、更大的界面和多价表位,共同阻断了传统意义上不可成药的 PPI。自 1990 年代以来,抗体稳步发展为一类具有临床意义的治疗实体,以免疫球蛋白 (IgG) 为原型。例如,曲妥珠单抗靶向HER2 用于治疗乳腺癌,而阿达木单抗结合并抵消TNF‑α 以抑制炎症和缓解类风湿性关节炎【2】。
最近,基于对肿瘤免疫检查点的研究,比如抑制PD‑1/PD‑L1的相互作用,产生了包括nivolumab和pembrolizumab在内的抗体治疗已经彻底改变了癌症治疗的方法。此外,抗体为不可成药的靶点的治疗提供了新策略。
抗体药物偶联物 (ADC),例如用于治疗 HER2 阳性乳腺癌的曲妥珠单抗emtansine,由三个部分组成:可以特异性结合抗原的作为锚定物的抗体,小分子药物有效载荷以发挥细胞毒性作用,以及连接器。
双特异性抗体,尤其是双特异性 T 细胞接合剂 (BiTE),能够靶向性激活自身T细胞杀伤肿瘤细胞。然而,基于抗体的药物有其固有的局限性,大分子量(IgG约 160 kD)和组织渗透性差大大减小了它们在细胞溶质药物靶点中应用的可能性。高效递送技术和小尺寸抗体模拟物(如 scFv 和纳米抗体)的发展可以提高它们在治疗不可成药靶点中的应用。
2.5 治疗性RNA技术
对于不可成药的靶标,另一种方法是在基因/转录水平而不是蛋白质水平上进行操作。小干扰 RNA (siRNA) 可以下调 mRNA和后续的蛋白质表达。已经有 50 多项基于 siRNA 的临床试验,包括尝试靶向突变的 p53 来治疗肾急性肾功能衰竭和沉默 cmyc 以治疗实体瘤或多发性骨髓瘤。
此外还有CRISPR技术,但是这些策略仍然面临着挑战,例如敲低效率和脱靶效应等。这些限制可以通过化学修饰、合理的序列设计和精确的RNA 载体系统来解决。基于 RNA 的疗法在治疗实体层面上可以与其它策略互补,以协同破解不可成药的靶标。
三、靶向不可成药靶点的药物发现技术
尽管多年来高通量筛选 (HTS) 促进了药物的发现,但一些缺点导致在靶向不可成药靶点方面的成功率较低,例如缺乏专为特定靶标设计的化合物库、长时间筛选和构建分子库的高成本。如果库中没有有效的治疗实体,则不会筛选出任何化合物,这对于针对缺乏明显配体结合口袋或类似物的不可成药靶点的治疗尤其重要。因此,考虑到不可成药靶点的特点,迫切需要开发新的药物发现技术(图4)。
3.1 生物展示技术
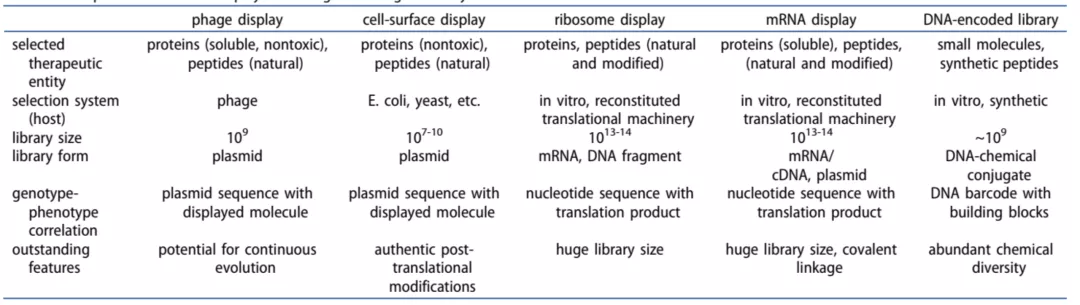
图4 药物发现中各种展示技术的比较
生物展示技术是开发高亲和力的生物分子(如蛋白质、 肽和核酸)的有效方法(图4)。较早的展示技术可以追溯到由噬菌体展示和细胞表面展示组成的细胞展示系统。表面展示是一种新的基因操作技术,主要原理是使表达的多肽以融合蛋白形式展现在核糖体、病毒或细胞表面,保持相对独立的空间结构和生物活性。
借以研究多肽的性质、相互识别和作用,筛选特定功能的多肽结构,实现蛋白质的固定化和定向进化。George P. Smith和George P.Winter因为"多肽和抗体的噬菌体展示技术”而获得2018诺贝尔化学奖。
细胞和无细胞(核糖体和mRNA)展示系统都提供丰富的序列多样性,并产生大量的治疗性蛋白质和肽。例如,组合抗体库的核糖体展示中产生的西妥昔单抗是一种靶向 EGFR 的嵌合单克隆抗体【3】。展示技术还用于研究不可药物靶标 KRAS 和 p53 的高亲和力结合的分子。
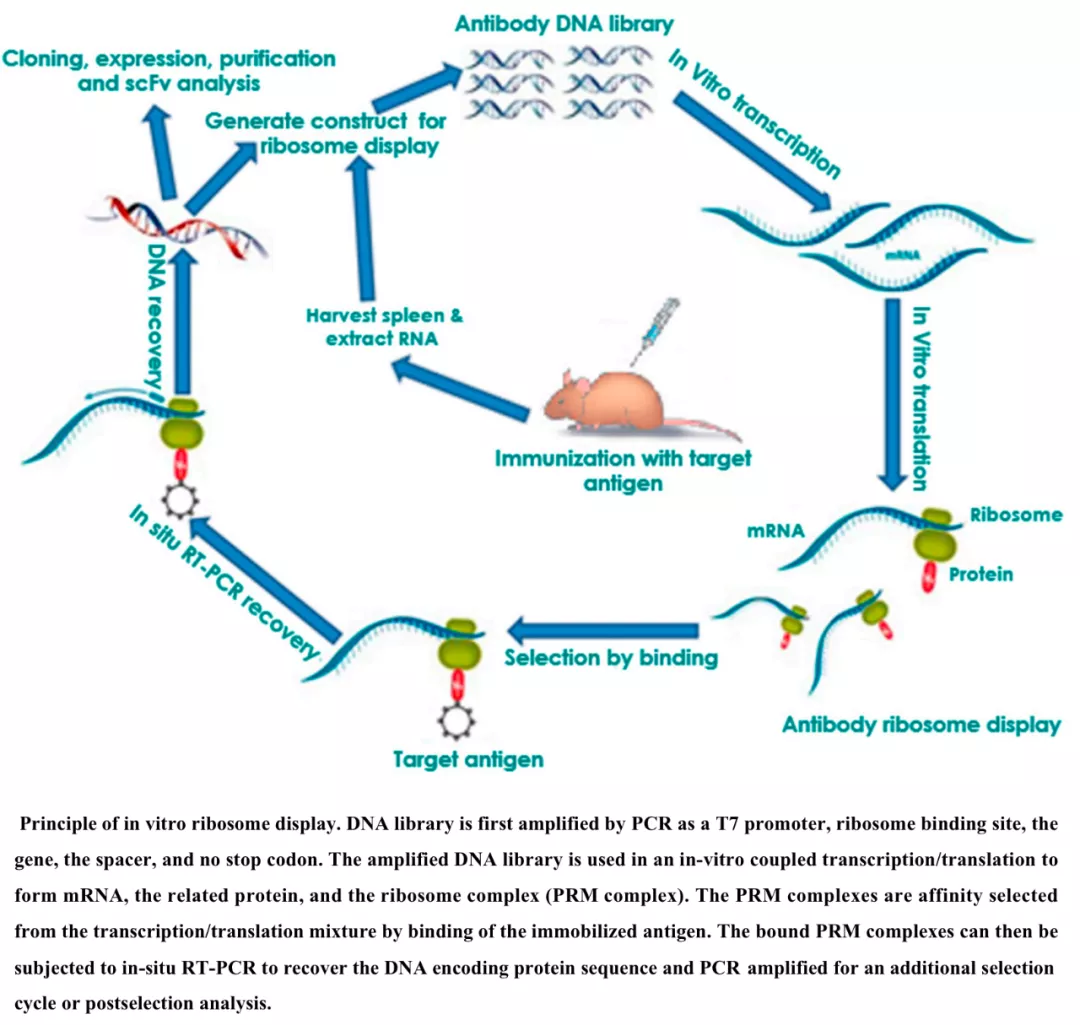
《Ribosome Display Technology: Applications in Disease Diagnosis and Control》
3.2 DNA编码文库 (DEL)
尽管生物展示技术有效地得到了基于蛋白质和基于肽的库,但它们的应用仅限于由天然(或受限)氨基酸构建块组成的治疗实体。值得注意的是,小分子药物和基于肽的药物包含更丰富的化学多样性,迫切需要被开发。DEL技术应运而生,它的优势在于两个方面:
首先,由于使用的构建模块和组合化学原理, 实现了丰富的化学多样性和提高了化合物库的容量。通过DEL开发的临床候选药物包括 GSK2256294, 一种可溶性环氧化物水解酶 (sEH) 抑制剂,用于治疗慢性阻塞性肺病 (COPD)和 GSK2982772,一种受体相互作用蛋白 1 (RIP1) 激酶抑制剂,用于治疗银屑病、类风湿性关节炎和溃疡性结肠炎。
其次,活细胞内基于 DEL 的选择可以在生理条件下捕获靶标特异性配体,针对内源性细胞表面蛋白的 DEL 选择避免了许多不可成药的膜蛋白的表达和纯化【4】。
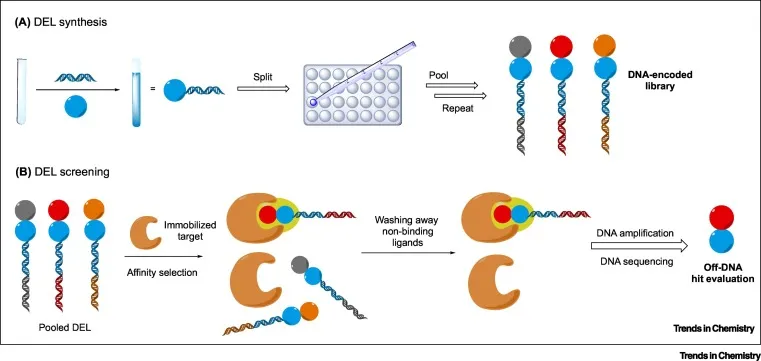
《Developments in Photoredox-Mediated Alkylation for DNA-Encoded Libraries》
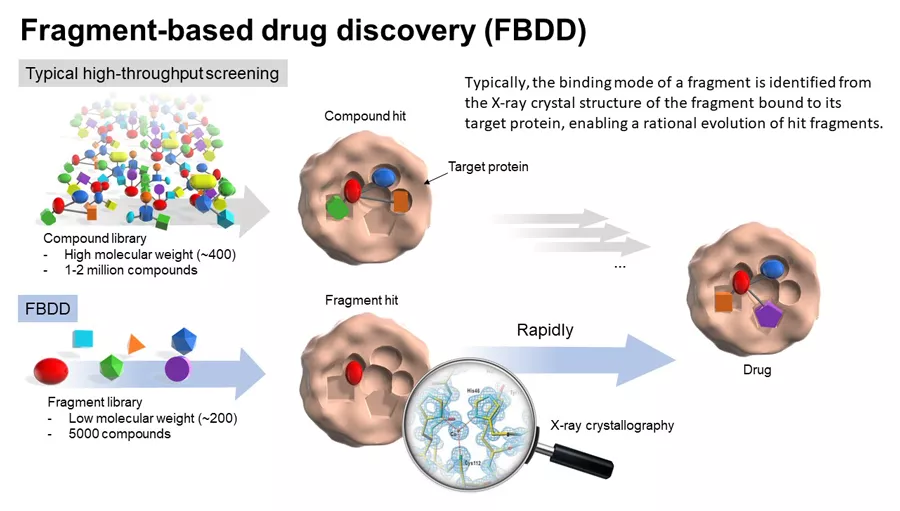
3.3 基于片段的药物发现
基于片段的药物发现 (FBDD) 不是像传统的 HTS 那样直接从庞大的集合中寻找药物大小的分子, 而是通过筛选活性片段,拼接设计合成化合物。 寻找片段的一般方法包括表面等离子体共振 (SPR)、 核磁共振 (NMR)、 X 射线晶体学和质谱 (MS)。FBDD 的优点如下:
首先,片段的简单结构使其更容易被挖掘出来,通常每次可以筛选出500‑1000个选择性片段,为后续优化提供更多选择。
其次,由于最佳的结合构象和方向,片段比大分子更有效地与目标结合。其中基于 FBDD设计合成的成功化合物有venotoclax,这是一种针对抗凋亡蛋白 Bcl‑2 的抑制剂,Bcl‑2 曾经被定义为不可成药的蛋白质,因为它的 PPI 界面平坦, 没有裂缝或配体袋。最近,共价片段药物发现也作为一种新方法出现。
3.4 变构位点的应用
并非所有蛋白质都为药物设计提供明显的配体结合位点(正构位点、活性位点)。这促使科学家寻找一些隐藏的变构位点(图5d)。变构位点显示出较少的保守性,提供了开发亚型选择性和低副作用抑制剂的可能性。
诺华曾设计出了第一个不可成药的靶标磷酸酶 SHP2 的变构抑制剂。作为一个蓬勃发展的领域, 变构抑制剂仍然存在若干科学挑战。POI 的整个晶体结构并不总是很清楚,从而阻碍了变构位点的解析。 此外,积累的化学物库不是为针对变构位点而设计的,从而降低了筛选过程中的成功率等。
3.5 虚拟筛选
虚拟筛选的明显优点是数据库的大小,它可以包含多达数万亿个虚拟化合物。虚拟筛选有两种方法,即基于结构的虚拟筛选(SBVS)和基于配体的虚拟筛选(LBVS),前者依赖于靶标的 3D 结构来运行对接过程进行结合模式预测,而后者主要侧重于药效团和亚结构的相似性搜索以及定量构效关系 (QSAR) 研究。
最近,SBVS 和LBVS 的结合已成为靶向不可药物靶点的有效方法。虚拟筛选可以在有限的时间内处理数百万个化学实体,从而显着降低早期药物发现的成本。(图5e)

图5 常见的不可成药靶标药物发现的技术方法
3.6 计算机辅助药物设计(CADD)
缺乏一些不可成药的靶标的 3D 结构阻碍了传统的药物发现。计算机辅助药物设计 (CADD) 是一种通过计算化学预测和计算结合模式的流行工具,与虚拟筛选方法的分类一样,CADD有两大类,即基于配体的药物设计(LBDD)和基于结构的药物设计(SBDD)。这里主要介绍SBDD,SBDD的关键在于明确了靶标结构。目前,预测或构建3D结构模型的主要方法有3种: (图5f)
- 同源建模,简而言之,是指以已知同源蛋白的实验3D结构为模板,对目标蛋白进行结构建模;
- 从头预测,与同源建模完全相反,是仅仅依靠一级序列来预测高级结构;
- 折叠识别,介于同源建模和从头预测之间。
RAS抑制剂的开发是将CADD策略应用于药物发现的案例之一,包括泛RAS抑制剂3144和BI2852。
四、总结
尽管人类基因组共编码了 20,000 多种蛋白质,但只有不到 1,000 种蛋白质被验证为药物靶点。基础生物学和医学研究仍在对疾病发生机制的进行更深刻的了解,通过进一步的研究,发现许多尚未成药或不可成药靶标与疾病的发生发展有密切的联系。例如,除了与细胞生长和增殖相关的经典药物靶点外,越来越多的证据表明免疫重编程、细胞代谢和细胞死亡途径都参与了肿瘤的发生。
总的来说,通过未来对疾病发生机制的深层次了解、治疗实体的创新和药物发现技术的进步,有望加速药物开发以解决“不可成药”问题并扩大可治疗疾病的范围。
参考资料
- Zhang J, Yang PL, Gray NS, et al. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9(1):28–39
- Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the high-hanging fruit.’ Nat Rev Drug Discov. 2018;17:197–22349
- Galán A, Comor L, Horvatić A, et al. Library-based display technologies: where do we stand?. Mol Biosyst. 2016;12(8):2342–2358.
- Huang, Y, et al. 2021. Selection of DNA-encoded chemical libraries against endogenous membrane proteins on live cells. Nature Chemistry 13, 77–88
- Gong Zhang, Juan Zhang, Yuting Gao, Yangfeng Li & Yizhou Li (2021):Strategies for targeting undruggable targets, Expert Opinion on Drug Discovery,
