【7.8.2】NGS质控质粒
一、Addgene 新的内部 NGS 流程
首先,该过程只能通过使用我们的高通量 DNA 分离过程来实现,该过程可产生足够数量的高质量分离 DNA 样本用于测序。该过程以平板形式完成,每周可生产 2 到 6 个平板,每块 96 个样品。这是我们的新流程将开始的地方。我们与 seqWell 合作,使用plexWell 技术轻松快速地创建 Illumina 测序文库。从开始到结束,文库制备过程只需要大约一天的时间,即使是六个板(576 个单独的质粒)!然后对这些文库进行 QC、合并并准备在我们新捐赠的 MiSeq 上进行 Illumina 测序。
对于完整的质粒测序,我们在 MiSeq 上执行 2x251 运行,大约需要两天才能完成。运行完成后,我们开始组装过程。再次感谢我们在 seqWell 的合作伙伴,我们使用了一个管道,该管道从我们池中的每个样本中获取原始数据,并将读数单独组装成我们的 QC 科学家可以轻松分析的单个 FASTA 序列(有关我们的注意事项,请参见下文)用于分析质粒序列时)。
与 Addgene 的所有事情一样,这个新流程的每个部分都涉及大量的 QC。首先,我们对每个分离的质粒样本进行 Picogreen 量化,并将每个样本归一化到 10 倍范围内。不符合此范围下限的质粒“失败”并从平板中取出以再次制备。然后这些空洞被我们队列中的其他样本填充,例如用于病毒基因组测序 (VGS) 的病毒样本。QC 在整个文库制备过程中进行,包括各种中间步骤的 Picogreen 量化、电泳凝胶的大小和最终文库的 qPCR 量化。
二、整个质粒的更广阔视野
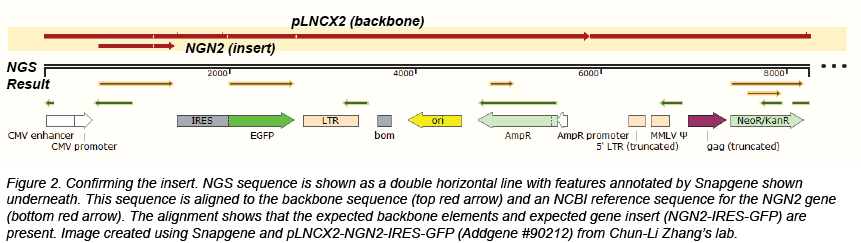
为了确认质粒的序列,我们检查三件事:
- 将 NGS 结果与参考序列比对以确认主干(backbone)元素。
- 通过对齐 NCBI 条目或使用 BLAST 来确认基因/插入。
- 确认标签和融合蛋白。
我们将在下面分解每个步骤。
2.1 对齐序列 Aligning the sequence
在对新保藏的质粒进行质量控制时,我们采取的第一步是将 NGS 结果与参考序列(如已知骨架序列)进行比对。我们不一定期望完美匹配 - 我们经常会在复制起点或其他常见主干元素中发现一些不匹配。由于我们已经成功地培养了质粒以进行测序,因此我们确信这几个小的错配通常不会影响质粒的功能。

2.2 确认插入 confirming the insert
我们通常通过BLAST或通过与 NCBI 参考序列或保藏人提供的序列直接比对来确认插入。保藏实验室通常会提供插入序列或带注释的 Genbank 文件,这些文件对更复杂的质粒有用,例如那些包含没有公开参考序列的合成区域或包含经过许多修饰的基因的质粒。我们寻找可能损害功能的点突变、截断和插入。当我们确实发现突变时,我们会检查它们是否影响翻译的氨基酸。我们还确认基因的种类与与质粒相关的数据相匹配。
确认标签和融合蛋白 confirming tags and fusion proteins
最后,我们通过使用 Snapgene 检测共同特征来确认启动子、标签、融合蛋白和可选择标记。如果我们发现信息与我们根据沉积实验室提供的数据所预期的不同,我们将这些称为“质量控制 (QC) 问题”。然后我们要求存放实验室检查差异。如果保藏实验室确认这些差异是预期的并且不影响质粒功能,我们将更新质粒页面上的信息。
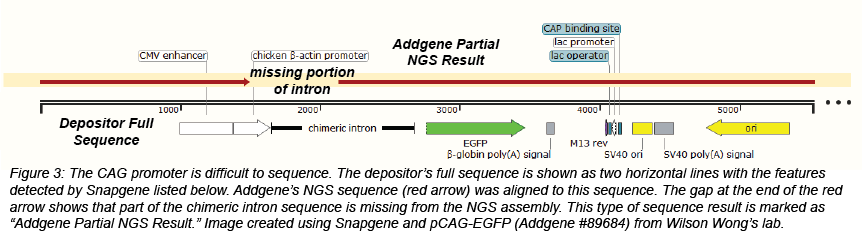
三、有时我们会丢失一部分质粒
某些质粒的某些区域特别难以测序和组装,包括富含 GC 的区域。例如,CAG 启动子,一种由 CMV 增强子、鸡 β 肌动蛋白启动子和兔 β-珠蛋白剪接受体组成的混合启动子,包含超过 80% GC 的序列。一些 IRES 序列还包含富含 GC 的区域。由于 NGS 将许多较小的序列组装成较大的序列,因此具有重复的区域可能无法正确组装。由于这些问题,某些质粒的 NGS 不会产生一个完整的循环组装。大多数情况下,这些质粒会作为一两个部分组件返回。如果我们获得有用的 NGS 结果,但不是 100% 完整,我们仍会将这些数据作为部分序列 提供,标题为“Addgene Partial NGS Result”。在某些情况下,质粒页面底部的保藏者评论部分会提供有关我们测序结果的更多信息。
四、病毒制备和汇集文库 Viral preps and pooled libraries
除了常规的质粒测序外,你们中的许多人都会熟悉我们为确保获得高质量、即用型病毒制备物和汇集文库所做的努力。
病毒样本被填充到与我们正常的完整质粒测序 (CPS) 工作流程相同的过程中,并产生足够的数据来捕获整个病毒基因组。该工作流程及其分析的完整细节可以在这里找到:一种新型的下一代测序和分析平台,用于评估来自病毒 DNA 提取物的重组腺相关病毒制剂的身份。
合并文库对 NGS 的 QC 更具挑战性,因为它们通常从一个文库到另一个文库大不相同。它们也不同于 CPS 过程,因为我们不会对池中的所有质粒进行完全测序,而是选择性地对池中质粒的可变部分进行测序 - 例如。CRISPR 文库中的 gRNA。一般而言,我们的 QC 科学家与保藏实验室合作开发适合相关文库的 PCR 策略。我们将订购可以放大文库插入片段的引物。此 PCR 步骤还添加了 Illumina 测序所需的接头序列以及每个样本的唯一条形码,以便我们可以将测序读数分配给正确的样本。然后执行 PCR,根据需要进行优化,并使用标准 PCR 净化试剂盒进行净化,以去除任何模板 DNA 和引物。
在对该样本执行大量 QC 后,我们会将其加载到我们的新 MiSeq 并设置运行,确保记下每个样本的适当条形码。大约 24 小时后,我们从仪器中卸载 FASTQ 数据并开始分析。我们使用Joung 等人描述的 Python 程序的修改版本。2017 年,我们希望很快将其作为我们 OpenBio 存储库的一部分提供。
参考资料
