【2.1.1】限制性克隆
一般都是黏性末端,即切口不是平的。
通过限制性消化和连接进行克隆时,您可以使用限制性内切酶切开质粒(骨架)并插入已被兼容的限制性内切酶切割的线性 DNA 片段(插入片段)。然后,DNA 连接酶将质粒与新片段共价结合,从而生成完整的环状质粒,可以轻松地在各种生物系统中维持。继续阅读以深入了解如何执行限制摘要。
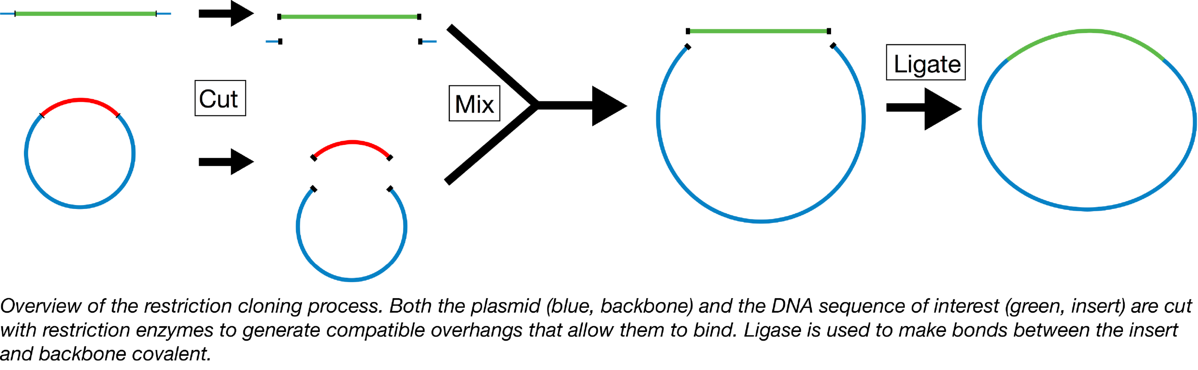
在开始限制性消化和连接过程之前,您应该仔细选择主链和插入片段 - 这两者都必须具有兼容的限制酶切割位点,以便您的插入片段以正确的方向放置到主链中。例如,如果您将基因克隆到表达载体中,您会希望基因的起点刚好位于骨架中的启动子下游。理想情况下,主链将包含启动子下游的各种限制性酶切位点(限制性位点),作为多克隆位点 (MCS) 的一部分。拥有多个位点可以让您轻松地根据启动子定位您的基因插入。
例如,假设您的质粒骨架类似于下图左侧的骨架。它有一个启动子(蓝色箭头),后跟限制性位点 EcoRI、XhoI 和 HindIII。要将您的基因放置在启动子下游的正确方向,您可以在基因开始的 5' 端添加一个 EcoRI 位点,在基因末端的 3' 端添加一个 HindIII 位点。这样你就可以用 EcoRI 和 HindIII 切割质粒骨架和插入片段,当你将切割的产物混合在一起时,两个 EcoRI 消化的末端会退火,两个 HindIII 消化的末端会退火,留下你的 5' 末端基因就在启动子的下游,并将基因放置在正确的方向。然后将连接酶添加到混合物中以共价连接主链和插入片段,PRESTO,您就有了一个完整的质粒,可以用于您的实验。

或者,如果您的插入片段两侧是该酶的限制性位点,则整个过程可以使用单个酶完成,但插入片段可以正向或反向与主链退火,因此您需要一些方法验证插入以您想要的方向结束 - 通常通过Sanger 测序或进一步的限制性消化。
1. 消化 Digestion
为您的插入片段(或供体质粒)和质粒骨架设置限制性消化。因为在凝胶纯化步骤中会丢失一些 DNA,所以消化大量起始材料很重要。我们建议使用 1.5-2μg 插入片段和 1μg 质粒骨架。用两种酶切割尽可能多的主链质粒也很重要,因此消化直到完成是很重要的。完全消化所需的时间因酶而异。许多公司现在销售可以在短短 10 分钟内消化大量 DNA 的快速消化酶,但请咨询您的酶制造商以确保您在适当的时间和条件下进行切割。
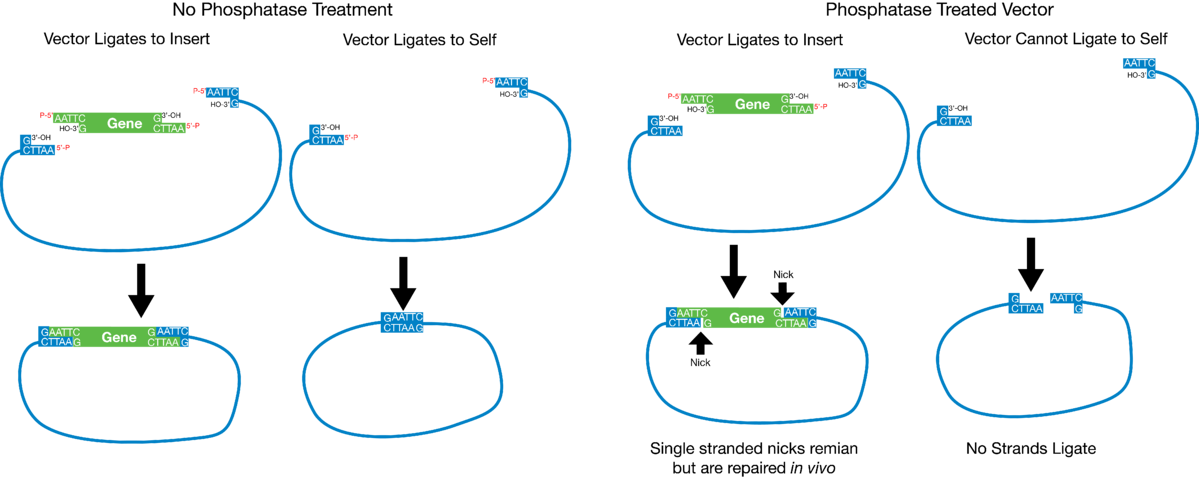
如果您打算仅使用一种限制性内切酶,或消化后具有相容突出端或无突出端的酶,则需要使用磷酸酶来防止主链质粒的重新环化(见下文)。根据您选择的磷酸酶,您应该在连接步骤之前或凝胶纯化步骤之前用磷酸酶处理您消化的骨架质粒。通常使用 CIP(小牛碱性磷酸酶)或 SAP(虾碱性磷酸酶)。按照制造商的说明进行操作。
2. 通过凝胶纯化分离您的插入片段和载体
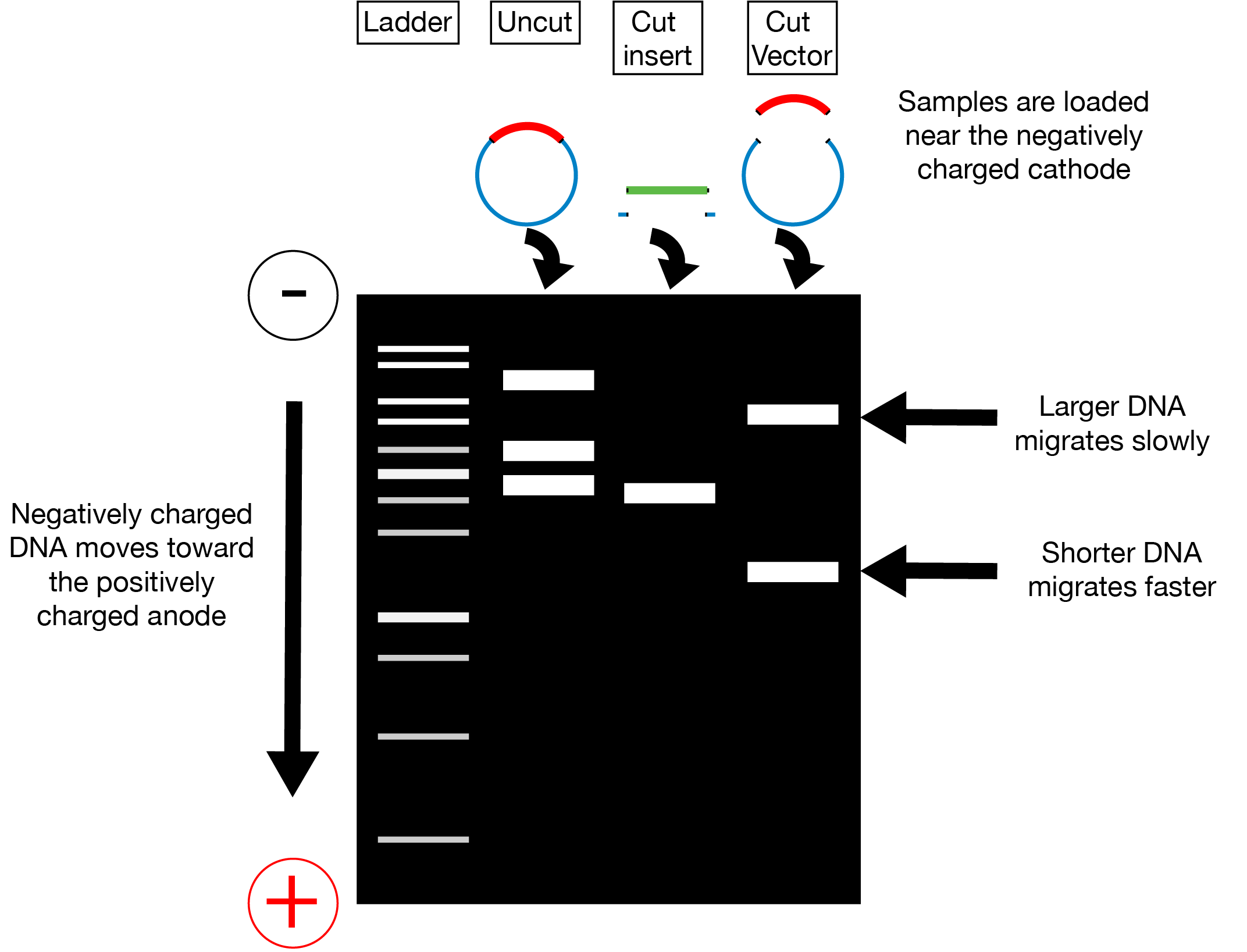
现在您已经切割了插入片段和载体,不幸的是,您不能将消化混合物放在一起。您需要将插入片段和骨架与用于消化它们的酶以及切下或切下的任何片段分离。一个简单的方法是凝胶纯化。在凝胶纯化中,您使用凝胶基质(通常是琼脂糖)上的电压差将带负电的 DNA 拉过凝胶。如左图所示,您消化的 DNA(和未消化的对照)装载在凝胶顶部,位于朝向阴极的孔中(- 电荷)。当在凝胶两端施加电压时,DNA 向阳极迁移(+ 电荷)。较大的线性化 DNA 片段比较小的线性化片段迁移得更慢。您可以通过不同的迁移速度将您的主干与从它切下的任何插入物以及新插入物与从它切下的任何悬垂物分开;在凝胶运行一段时间后,这些不同大小的碎片将位于不同的位置,可以单独从凝胶中切下。
有多种方法可以显示凝胶中的 DNA(此表不包括所有凝胶染色):
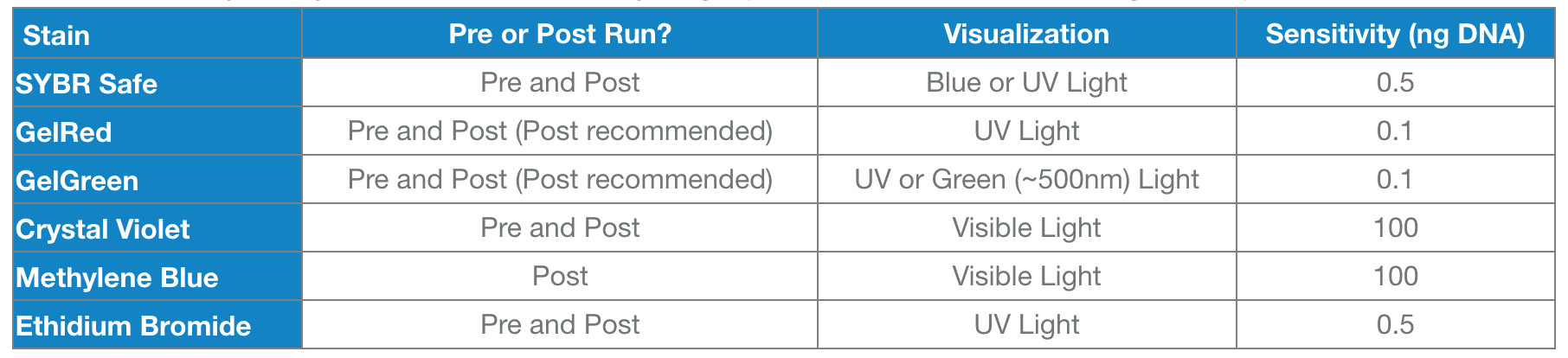
有关这些stains的更多信息,请参阅Bitesize Bio 博客 及其相关制造商网站。
这些污渍(stains)需要您在运行您的样品后,要么弄脏(stain)您的凝胶或添加染色的凝胶正在取得(后或前在上表中运行,分别)。上述一些污渍需要您使用紫外线来观察您的 DNA - 请注意,紫外线会损坏 DNA,并且在使用紫外线进行可视化时应佩戴适当的个人防护设备,因为它会损坏眼睛和皮肤。
为纯化目的运行凝胶时,重要的是要有漂亮的清晰条带并留出空间来剪掉条带。因此,我们建议您使用较宽的凝胶梳,在较慢的一侧运行凝胶,并在样品之间跳过泳道。除了 DNA 阶梯标准外,如果您的消化不符合预期,运行每个质粒的未切割样品也是一个好主意,以帮助排除故障。
通过您最喜欢的凝胶纯化方法从凝胶上切下并纯化插入片段和受体质粒骨架带后,确定回收 DNA 的浓度很重要,因为这对连接步骤非常有用。
3. 将您的插入物结扎到您的载体中 Ligate Your Insert into Your Vector
在连接步骤中,您将纯化的、切割的主链和插入物混合在一个管中,使限制性消化产生的相容突出端彼此退火并形成完整的环状质粒。然后添加 DNA 连接酶以牺牲 ATP 为代价将片段共价连接在一起(见下文,共价键以红色表示)。

我们建议在标准连接反应中使用大约 100 ng 的总 DNA。理想情况下,您需要大约 1:3 的“受体质粒:插入比例”。由于每个碱基对的数量各不相同,因此很难仅根据 DNA 浓度来计算。一种方法是对您尝试创建的每个质粒进行 2 次连接,使用不同比例的受体质粒插入。
设置不添加插入 DNA 的阴性对照连接反应至关重要。这将允许您确定由于背景再环化和未切割质粒的污染,您应该在转化中预期多少个菌落。
4. Transformation
将您的连接反应Transformation为您选择的细菌菌株。按照制造商对感受态细胞的说明进行操作。
对于大多数标准克隆,您可以将 1-2μl 的连接反应 transform 为感受态细胞,例如 DH5alpha 或 TOP10。如果使用更少的总 DNA (<1ng) 或者如果您无法获得集落,您可能需要使用更高的感受态细胞。此外,如果您的最终产品将非常大 (>10kb),您可能需要使用电感受态细胞而不是更常见的化学感受态细胞。
您应该在ligation后至少执行两次转换:
- 含有单独骨架的连接混合物的对照转化;
- 含有插入物和骨架的连接混合物的转化。
指示成功和不成功连接的样品结果如下所示。成功的连接将在仅骨干板上有很少的菌落,而在骨干 + 插入板上有许多菌落(或至少比单独骨干板上的菌落更多)。不成功的连接通常会导致两个平板上的菌落很少(不成功 1),在单独的载体板中菌落比载体 + 插入板(不成功 2)多得多,或者每个板上的菌落数量大致相等(不成功 3)。
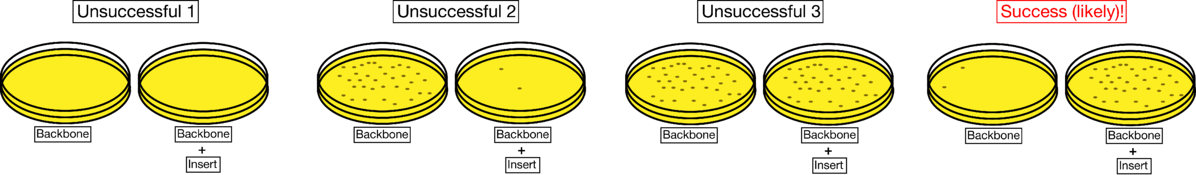
如果你有你的骨干板高菌落数(大于或等于骨干+插入,ü nsuccessful 2和3段),你可以尝试在存在和不存在连接酶单独结扎收件人(recipient)质粒。如果集落是由未切割的空质粒产生的,那么当您不添加连接酶时,您仍然会有集落。如果集落是受体质粒自连接的结果,当您添加连接酶时,您会看到明显更多的集落。
如果您没有看到任何菌落,您应该进行阳性对照以确保您的转化有效。您还应该验证您是否接种了合适的抗生素,并尝试在连接反应中改变“受体质粒:插入比例”(“recipient plasmid : insert ratio” )
5. 纯化完成的质粒 Purify the finished plasmid
一旦看起来您的结扎(ligation)有效,您将需要挑选单个细菌菌落并检查它们是否成功结扎。根据对照板上的背景菌落数量选择 3-10 个菌落(背景越多,您需要选择的菌落越多)并培养过夜培养物以进行 DNA 纯化。您可以进行的最简单的纯化是小量提取,但如果您需要大量 DNA,则需要进行中量提取或大量提取。在这些纯化中,您通常会裂解细菌;添加化学物质以沉淀出高分子量基因组 DNA;通过结合质粒DNA并让其他材料通过的柱过滤剩余的质粒DNA;最后,使用特定的缓冲液或水从柱子上选择性地洗脱质粒 DNA。有关更多详细信息,请参阅色谱柱制造商。寻找我们网站上的无柱纯化方案。
6. 验证质粒
通过限制性消化进行质粒克隆验证经过纯化的DNA,进行诊断限制性消化与您用于克隆的酶纯化的DNA 100-300ng的。在琼脂糖凝胶上运行消化。您应该会看到两条条带,一条与您的骨干尺寸相同,另一条与新插入件的尺寸相同(见右图)。如果您仅使用一种酶或使用具有兼容突出端的酶进行连接,则您需要验证插入片段的方向。您可能需要为此目的设计诊断摘要。
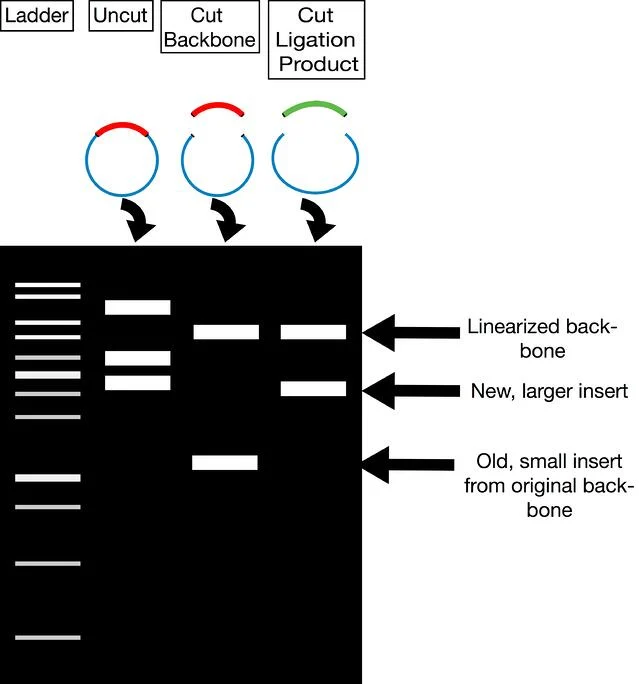
理想情况下,一旦您知道您的质粒具有适当大小的插入片段,您应该将其发送到使用允许您阅读插入片段的引物进行桑格测序。有关更多详细信息,请参阅我们关于如何验证您的质粒的帖子。
一旦您的完整质粒得到验证,您就可以开始实验了
参考资料
