【5.1.1】分子对接
一、基本原理
分子对接是直接药物设计的重要方法之一,其本质是两个或多个分子之 间的识别过程,涉及分子之间的空间匹配和能量匹配;最初思想起源于“锁- 钥模型” (Lock-and-key theory),即一把钥匙开一把锁
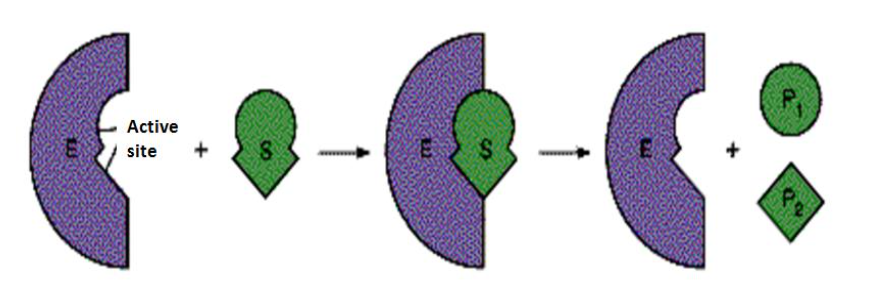
分子对接计算就是将配体小分子放置于受体的活性位点,并寻找 其合理的取向和构象,使得配体和受体的形状和相互作用的匹配最佳
分子对接方法分类
| 对接类型 | 方法特点 | 计算量 | 应用 |
|---|---|---|---|
| 刚性对接 | 参与对接的分子构象不发生变化,;仅改变分子的空间位置与姿态 | 简化程度最高,计算量相对较小 | 适合于大分子之间的对接 |
| 半柔性对接 | 允许对接过程中小分子构象发生一 定程度的变化,但通常会固定大分 子的构象,另外小分子构象的调整 也可能受到一定程度的限制,如固 定某些非关键部位的键长、键角等 | 兼顾计算量与模型的预测能力 | 应用比较广泛 |
| 柔性对接 | 允许研究体系的构象发生自由变化 | 计算量非常大,消耗机时很多 | 适合精确考察分子间的识别 |
分子对接常用软件
常用于分子对接的软件包括:
CDOCKER(Discovery Studio)、GOLD、AutoDock、Glide(Maestro)等
CDOCKER 采用半柔性的对接方法,可以产生高精度的对接结果
所需功能和模块:Discovery Studio Client /Macromolecules /Small Molecules /Receptor-Ligand Interactions
实验内容
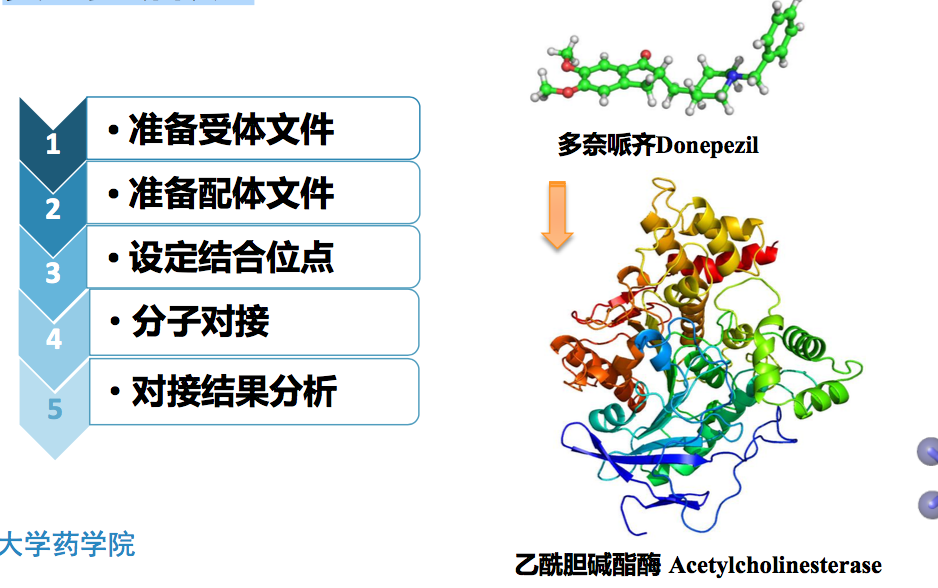
使用DS中CDOCKER模块将多奈哌齐药物分子对接至其作用靶标乙酰胆 碱酯酶的活性口袋中,找出两者结合的最佳构象状态,并分析相互作用。
所需数据文件:
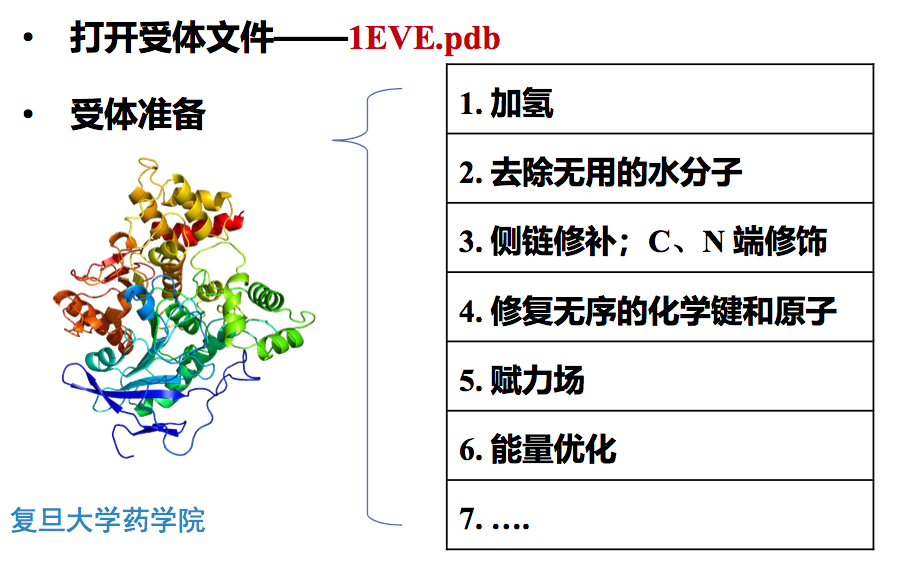
晶体复合物pdb文件(1EVE.pdb) ——Acetylcholinesterase + Donepezil (Aricept)
配体mol2文件(Donepezil.mol2)
二、分子对接 实验操作
实验步骤概述
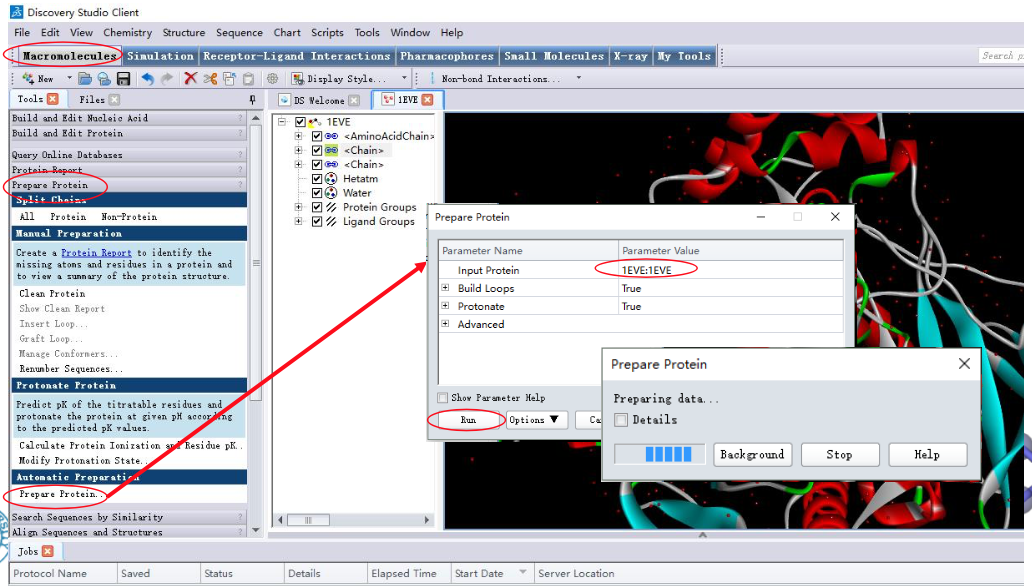
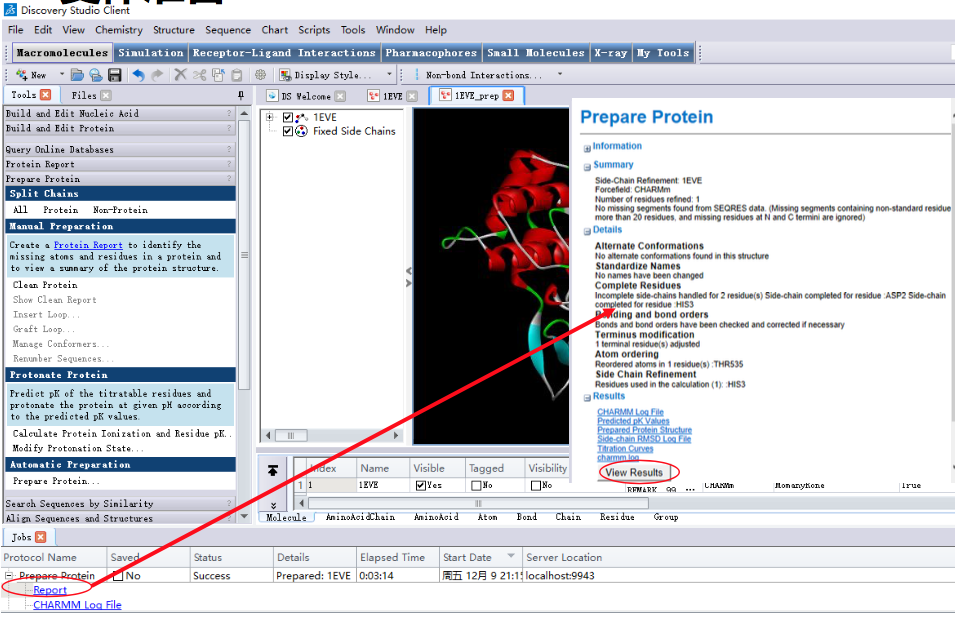
2.1 受体准备
2.2 配体准备
打开配体文件( Donepezil.mol2 )
- 改变离子化状态
- 产生互变异构体
- 产生立体异构体
- …

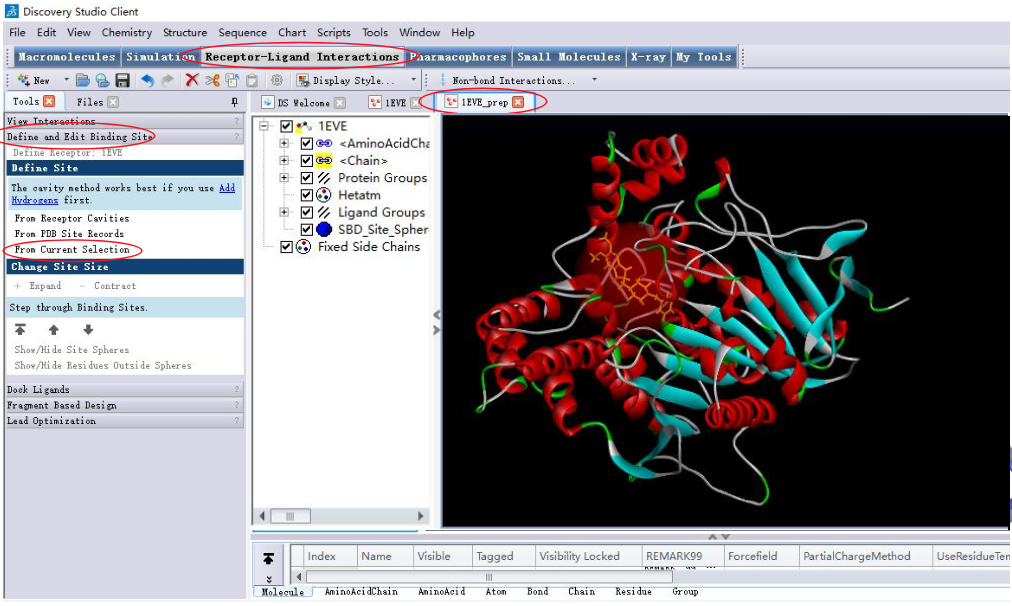
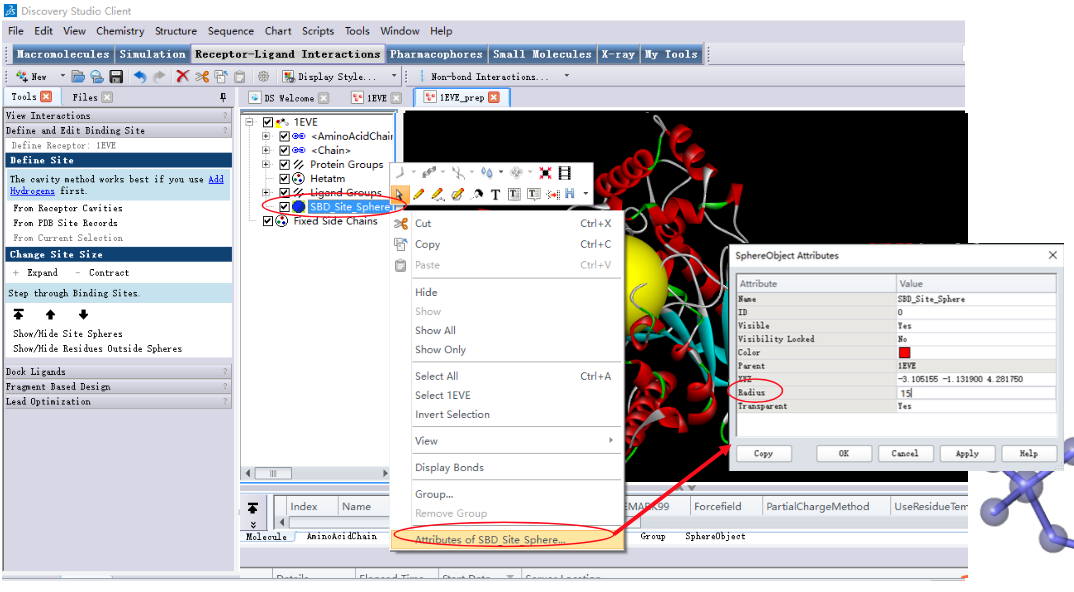
2.3 设定结合位点
结合位点的确定对于分子对接结果准确性的影响很大,因为配体分子 与受体相互作用过程的模拟主要是参考结合位点的几何特征进行的。
产生结合位点的方法:
- 以原配体的位置为中心产生结合位点;
- 参考文献,以活性口袋周围的关键残基为中心产生结合位点;
- 根据活性口袋特征探测活性位点。
2.4 分子对接

2.5 结果分析
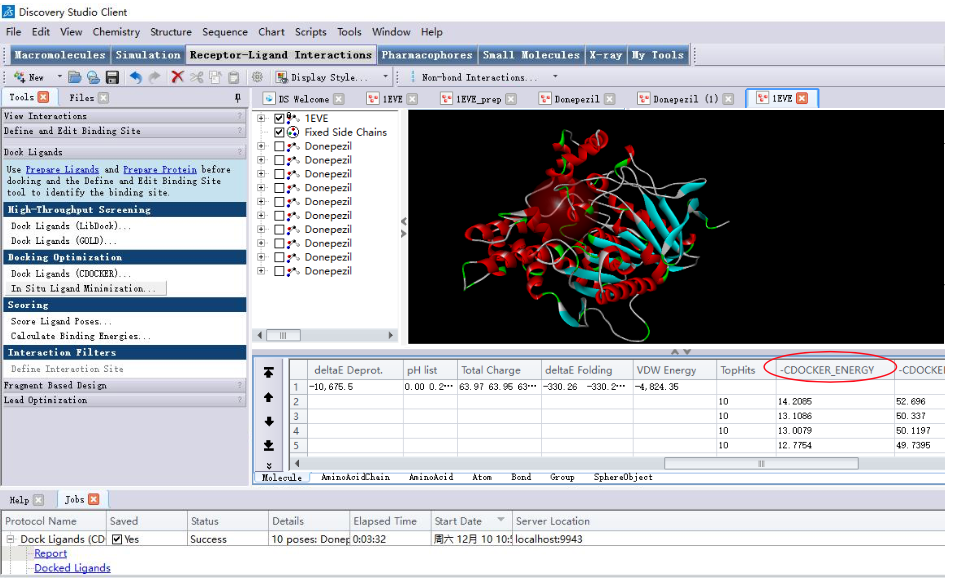
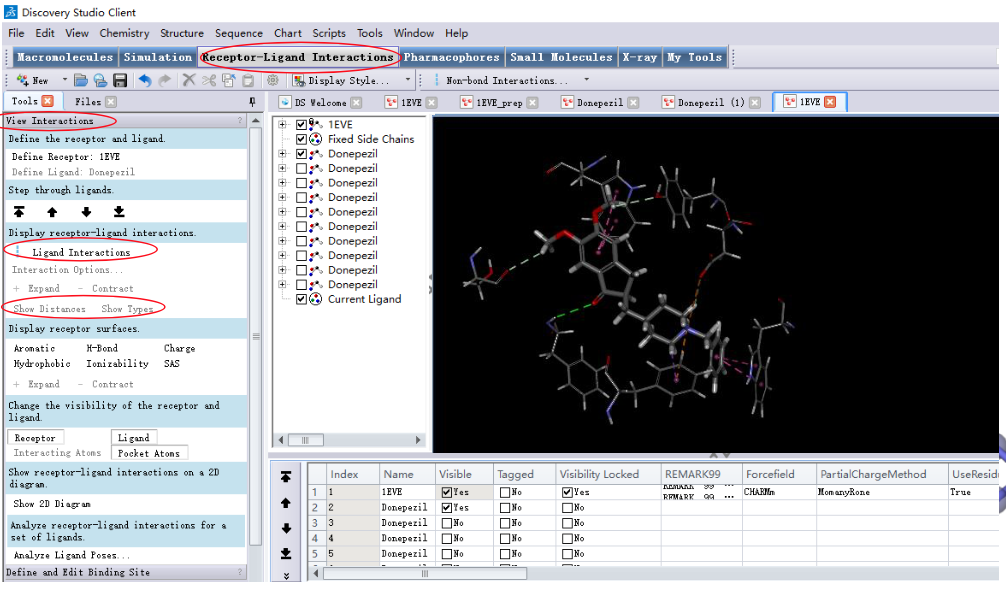


2.6 对接构象选取
- 复合物的结合能 / 相互作用力的强弱
- 对接结果出现的频率
- 实验数据是指导构象选择的重要依据
参考资料
- 复旦大学 李炜 老师的 《药物设计学》 课件
这里是一个广告位,,感兴趣的都可以发邮件聊聊:tiehan@sina.cn
![]() 个人公众号,比较懒,很少更新,可以在上面提问题,如果回复不及时,可发邮件给我: tiehan@sina.cn
个人公众号,比较懒,很少更新,可以在上面提问题,如果回复不及时,可发邮件给我: tiehan@sina.cn
